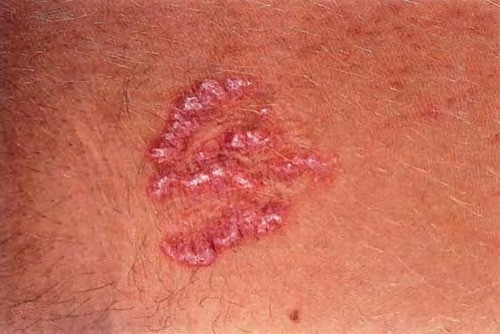
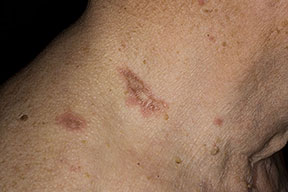
Elastosis Perforans
Serpiginosa
EPS is a unique perforating disorder characterized by transepidermal elimination of elastic fibers. The distinctive lesions are serpiginous in distribution and can be associated with specific diseases.
HISTORY
ETIOLOGY AND PATHOGENESIS
D-penicillamine appears to be a clear trigger for EPS. Alterations of elastic fibers have been described in patients with penicillamineinduced EPS, and this condition has usually occurred after long-term therapy of Wilson disease or cystinuria. EPS has also been reported in a child treated with D-penicillamine for juvenile rheumatoid arthritis. Abnormal elastic fibers have been found in the lungs as well as the skin of penicillamine-treated patients. Abnormalities of collagen have also been described in patients with EPS. The precise mechanism by which penicillamine induces EPS is not entirely known, but the drug chelates copper, the co-factor for lysyl oxidase, which is the enzyme that cross-links elastin.
Although most cases are sporadic, familial cases of EPS have been reported with different modes of inheritance. In at least one family, autosomal dominant inheritance with variable expression of EPS was suggested. Several inherited disorders of connective tissue have also been associated with EPS , suggesting that various pathways can lead to the abnormal elastin that results in this condition.
CUTANEOUS LESIONS
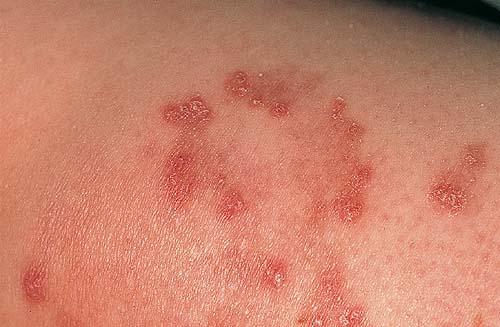
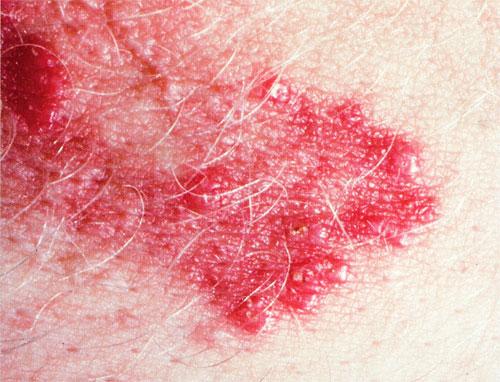
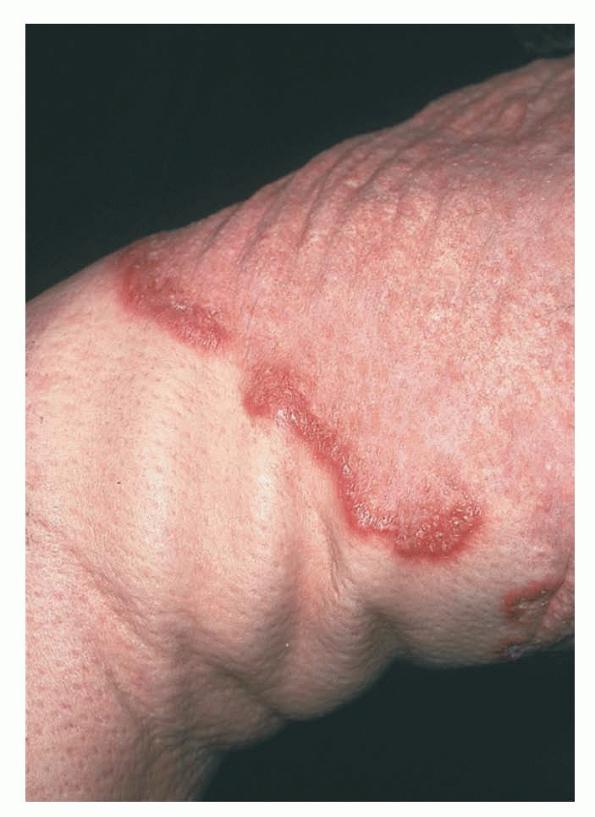
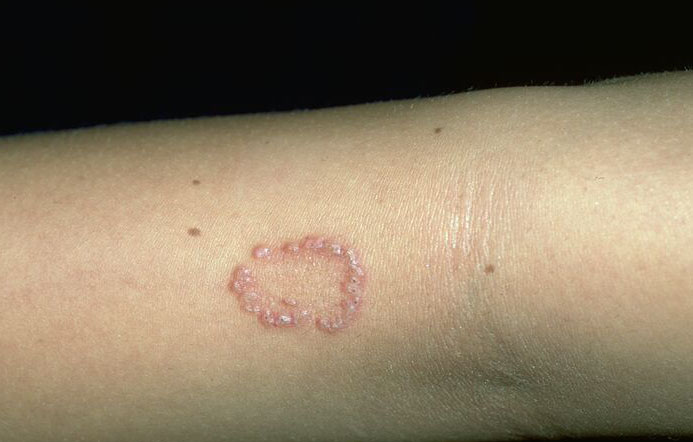
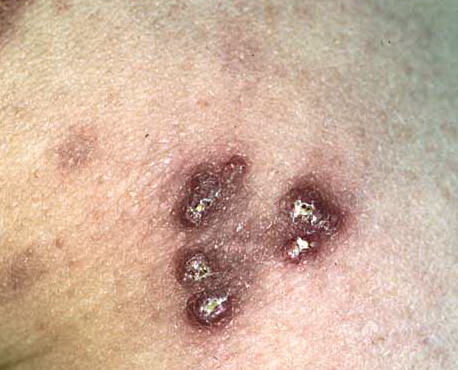
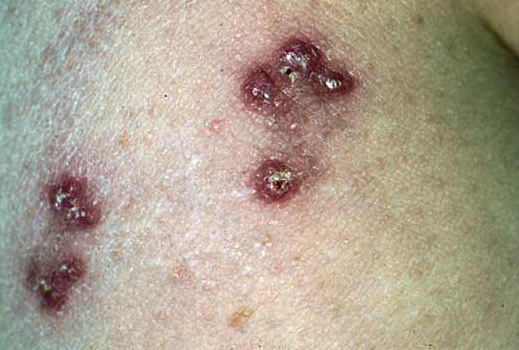
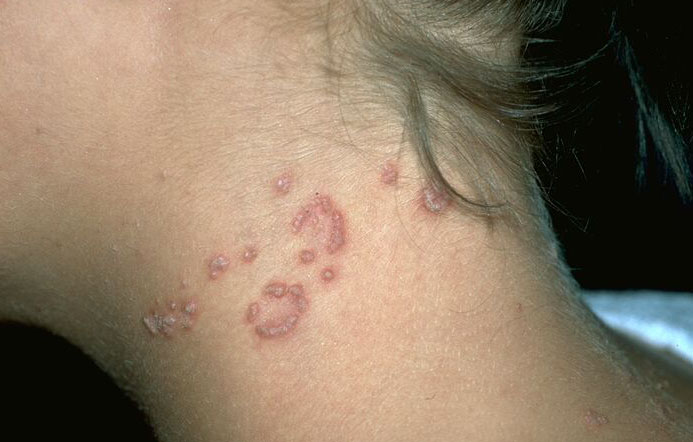
Skin lesions consist of hyperkeratotic papules ranging from 2 mm to 1 cm in diameter in a serpiginous distribution . Skin within these serpiginous arcs is often atrophic. The most common site of involvement is the neck , but lesions can also occur on the extremities and, more rarely, on the trunk. In rare instances, elastic tissue in the endocardium, bronchiolar walls, and arteries is involved. Rupture of the aorta has been reported.
RELATED PHYSICAL FINDINGS
EPS is associated with numerous inherited disorders, including osteogenesis imperfecta, cutis laxa, acrogeria, Ehlers-Danlos syndrome, Marfan syndrome, Rothmund-Thomson syndrome, and Down syndrome. EPS has been reported in association with administration of D-penicillamine to treat Wilson disease and with co-existing acquired cutis laxa after D-penicillamine therapy of cystinuria.
HISTOPATHOLOGY
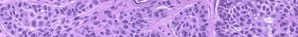
Histologically, lesions are characterized by transepidermal elimination of elastic fibers. Typical changes can be identified and consist of an inflammatory response around abnormal elastic tissue in the papillary dermis. The elastic tissue penetrates through channels in the acanthotic epidermis. The channels may be straight or tortuous, and there may be multinucleated giant cells in the surrounding inflammatory infiltrate. In addition to elastic tissue, the channels contain necrobiotic material with degenerated epithelial cells and parakeratotic nuclei of inflammatory cells. The top of the epidermal channels is also filled with keratotic material.
▪ DIFFERENTIAL DIAGNOSIS
The one entity easily confused with perforating diseases is prurigo nodularis. Other perforating disorders, such as perforating pseudoxanthoma elasticum (PXE) and perforating granuloma annulare, are easily distinguished from the acquired perforating disorders by skin biopsy.
Box 67-1 Differential Diagnosis of Acquired Perforating Disorders
- Perforating pseudoxanthoma elasticum
- Prurigo nodularis
- Excoriated dermal diseases (e.g., granuloma annulare, lichen planus)
- Hyperkeratosis follicularis perstans (Flegel disease)
- Acquired perforating dermatosis
Treatment for Acquired Perforating Disorders
|
|
TOPICAL
|
PHYSICAL
|
SYSTEMIC
|
|
First line
|
Corticosteroids
|
Broadband ultraviolet B
|
Acitretin, 30 mg daily
|
|
|
|
Narrowband ultraviolet B
|
Isotretinoin, 1 mg/kg in two divided doses daily
|
|
Second line
|
Retinoids
|
Psoralen and ultraviolet A light
|
Doxycycline, 100-200 mg daily in two divided doses; allopurinol, 100 mg daily
|
|
|
|
Surgical removal combined with oral clindamycin
|
EPS is easily distinguished from other perforating disorders when characteristic serpiginous lesions are present (in contrast to individual papules in the latter) and by the finding of elastic fibers in the perforating tissue on histology. In contrast to the other perforating dermatoses, which are usually associated with diabetes or renal failure, EPS has an entirely different group of disease associations as mentioned in Elastosis Perforans Serpiginosa. There is a single case report of EPS in a patient with renal disease. Histologically, however, the perforating elastic fibers of EPS usually allow differentiation, although fragments of elastic fibers may be found in the necrobiotic plug of other perforating disorders. At least one study reports the transepidermal elimination of elastic fibers in follicular occlusion disease, distinguishing that disorder from EPS.
Individual lesions of perforating PXE can simulate those of the acquired perforating dermatoses, but patients with PXE have characteristic yellow papules and plaques on the neck, axillae, and other flexural sites . Moreover, on skin biopsy, von Kossa stains show calcification of elastic fibers, and Verhoeff-van Gieson stains show fragmentation and clumping of elastic tissue. There have, however, been cases of PXE with serpiginous perforating papules containing calcified elastic fibers, thus simulating EPS. The lesions of perforating granuloma annulare can also simulate acquired granuloma annulare, but on biopsy, palisading granulomas are seen . Flegel disease (hyperkeratosis follicularis perstans), a rare autosomal dominant genodermatosis, is also characterized by keratotic papules, but these are smaller and also involve the lower legs. Histologically, Flegel disease is not a perforating condition, but, occasionally, it has been confused with perforating disorders.
▪ PROGNOSIS, CLINICAL
COURSE, AND TREATMENT
The perforating disorders are difficult to treat . Their prognosis is tied to the underlying diseases, so treatment of the underlying cause can be effective. Although some have reported spontaneous disappearance of perforating disorders with stabilization of renal disease, most of the perforating diseases continue for years unless treated. Perforating disorders frequently resolve after renal transplantation in patients with underlying kidney failure. Although there are isolated reports of successful therapy using topical retinoids, oral retinoids, oral antibiotics (doxycycline and clindamycin), and allopurinol, these are not usually effective. Phototherapy with narrowband ultraviolet B (UVB) or broadband UVB can result in substantial improvement, but is not as reliably effective as phototherapy for uremic pruritus. UV light treatment, if successful, tends to require sessions two to three times per week with 10 to 15 exposures for clearance. Many of the treatments that have been applied to the other perforating disorders have also been used for EPS. Isotretinoin and destructive modalities, such as cryotherapy70 and lasers, have also been used effectively for EPS.