Cockayne Syndrome
(Including Xeroderma Pigmentosum-Cockayne Syndrome Overlap)
CS is a very rare, autosomal recessive degenerative disease with cutaneous,
ocular, neurologic, and somatic abnormalities A review published in 1992 described 140 cases reported in the literature.
CLINICAL FEATURES
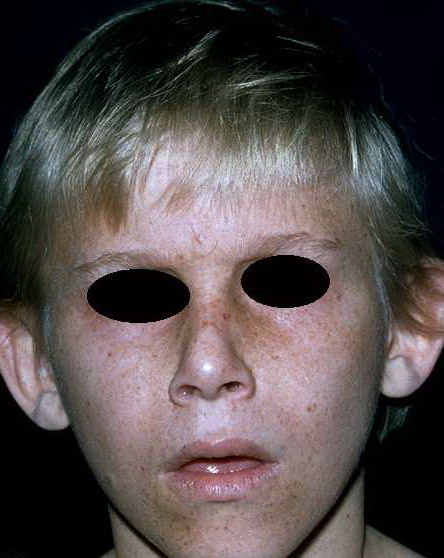
In 1936, E.A. Cockayne described a syndrome characterized by cachectic dwarfism, deafness, and pigmentary retinal degeneration with a characteristic “salt and pepper” appearance of the retina. The skin had photosensitivity without the excessive pigmentary abnormalities seen in XP. There was marked loss of subcutaneous fat, resulting in a “wizened” appearance with typical “bird-headed” facies and prominent “Mickey Mouse” ears. Other ocular findings included cataracts and optic atrophy. Neurologic abnormalities, in addition to deafness, include peripheral neuropathy, normal pressure hydrocephalus, and microcephaly. Birth weight and early development are usually normal. The disease onset is usually in the second year of life with slowly progressive neurologic degeneration. Intellectual deterioration may be non-uniform, with some functions preserved better than others. A severe infantile form has been described as well as a milder form with late onset. COFS syndrome with microcephaly and severe mental retardation and CAMFAK syndrome of congenital cataracts, microcephaly, failure to thrive, and kyphoscoliosis73 have some similar features. CS is not associated with an increased incidence of neoplasia.
LABORATORY ABNORMALITIES
Clinical laboratory testing often shows sensorineural deafness, neuropathic electromyogram, and slow motor nerve conduction velocity The electroencephalogram may be abnormal, and x-ray examination may show thickened skull and microcephaly. Computed tomography may be diagnostically useful in the detection of normal-pressure hydrocephalus and showing calcification of the basal ganglia and other structures . MRI of the brain shows atrophy and dysmyelination of the cerebrum and cerebellum. Bone age is usually normal. Height and weight are usually well below the third percentile for the age.
Cellular Hypersensitivity.
As with XP, cultured cells (fibroblasts or lymphocytes) from patients with CS are hypersensitive to UV-induced inhibition of growth and colony-forming ability. Host cell reactivation of UV-damaged adenovirus or plasmids is reduced, although generally to a lesser extent than in XP. There are two complementation groups (A and B) in CS . Patients with defects in CSA or CSB have similar features. A patient with COFS syndrome was reported with a defect in CSB73 and another with a defect in XPD.
Prenatal Diagnosis.
Prenatal diagnosis of CS has been performed based on the delay in recovery of post-UV RNA synthesis and the increased cell killing by UV radiation.
Xeroderma Pigmentosum-Cockayne Syndrome Complex
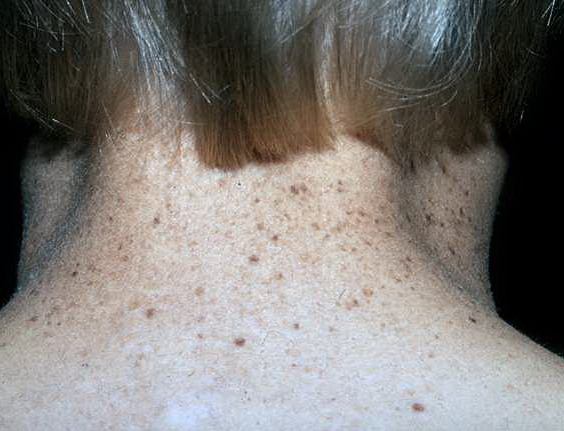
Approximately one dozen patients with CS have been found to have, in addition, clinical features of XP. These features include freckling on sun-exposed skin and cutaneous neoplasms . Cells from these XP-CS patients have reduced DNA excision repair characteristic of XP. Clinically, these patients may be distinguished from XP patients with neurologic abnormalities by the presence of the CS features of pigmentary retinal degeneration, calcification of the basal ganglia, normal-pressure hydrocephalus, and hyperreflexia. Cells from patients with this complex have been found to have mutations in three different XP genes: XPB, XPD, and XPG, demonstrating that several different genes are implicated in this disorder .
Trichothiodystrophy
TTD is an autosomal recessive disorder that is characterized by sulfur deficient, brittle hair and includes a spectrum of clinical phenotypes that may include photosensitivity, ichthyosis, intellectual impairment, short stature, microcephaly, characteristic facial features (protruding ears, micrognathia), and decreased fertility Some patients have immunodeficiency, dystrophic nails, and cataracts. Developmental delay may be associated with dysmyelination, a feature similar to CS; however, patients do not have retinal changes of CS. The spectrum of clinical involvement is broad ranging from only hair to severe multisystem abnormalities . TTD encompasses patients who have been described as Amish brittle hair syndrome, Sabinas brittle hair syndrome, or Pollitt syndrome.
In 1980, Price proposed the term trichothiodystrophy [derived from Greek tricho (hair) thio (sulfur) dys (faulty) trophe (nourishment)] recognizing the hair shaft sulfur deficiency as a marker for this symptom complex. Under light microscopy with polarization, TTD hair shafts have a characteristic dark and light banding pattern which gives a “tiger tail” appearance (see Fig. 140-6E). In addition, they usually have hair shaft abnormalities including trichoschisis (a clean transverse break through the hair) , trichorrhexis nodosa-like defects, and ribboning Hair shafts are brittle because of a reduction of high-sulfur matrix proteins, and amino acid analysis of the hair demonstrates reduced levels of cysteine and cystine in hair shaft proteins. This is associated with a decrease in the ratio of strong to weak disulfide bonds within the hair shafts.
Approximately 50 percent of TTD patients have clinical photosensitivity that ranges from subtle to severe. However, in contrast to the typical clinical features of XP, patients with TTD do not develop poikilodermatous changes (hyper- and hypo-pigmentation, telangiectasias, and atrophy) or skin cancers. Rarely, patients may have features of both TTD and XP, with typical hair features of TTD and the pigmentary and skin cancers characteristic of XP .
The majority of TTD patients have a defect in XPD (ERCC2). A few have mutations in XPB (ERCC3) or TTDA (TFB5), genes which are components of the transcription factor TFIIH that regulates both DNA repair and transcription. Some TTD patients have mutations in TTDN1, a gene of unknown function.85 It is believed that mutations that affect the repair function of the NER genes are associated with features of XP, whereas mutations affecting the transcription-related function results in features of TTD.
The diagnosis of TTD is based on examination of hair shafts . TTD hair shafts display tiger tail banding under polarizing microscopy. In addition, they show a spectrum of typical hair shaft abnormalities, including trichoschisis, trichorrhexis nodosa-like fractures, surface irregularities, and ribboning. Amino acid analysis of hair shafts can confirm reduced levels of cysteine and cystine.
Management includes sun protection and varies with the individual clinical features. Patients with developmental delay and intellectual impairment may have dysmyelination as seen on MRI of the brain and may benefit from neurologic and developmental assessment. Ophthalmologic involvement can include cataracts (which may be congenital), nystagmus, and errors of refraction. Some patients have recurrent infections that have been managed with prophylactic antibiotics or intravenous immunoglobulin G. Patients with skeletal abnormalities may benefit from rehabilitation medicine evaluation and support