Amyloidosis
Systemic amyloidosis can be classified as follows: (1) primary systemic amyloidosis (PSA), usually with no evidence of preceding or coexisting disease, paraproteinemia, or plasma-cell dyscrasia; (2) amyloidosis associated with multiple myeloma; or (3) secondary systemic amyloidosis with evidence of coexisting previous chronic inflammatory or infectious conditions.
Primary systemic amyloidosis involves mainly mesenchymal elements, and cutaneous findings are observed in 30-40% of patients. Secondary systemic amyloidosis does not involve the skin, whereas localized amyloidosis does.
Primary systemic amyloidosis involves the deposition of insoluble monoclonal immunoglobulin (Ig) light (L) chains or L-chain fragments in various tissues, including smooth and striated muscles, connective tissues, blood vessel walls, and peripheral nerves.1 The amyloid of primary systemic amyloidosis is made by plasma cells in the bone marrow. These L-chains are secreted into the serum. Unlike the normal L-chain and the usual form seen in patients with myeloma, these L-chains are unique in that they undergo partial lysosomal proteolysis within macrophages, and they are extracellularly deposited as insoluble amyloid filaments attached to a polysaccharide. Sometimes, instead of an intact L-chain, this amyloid has the amino-terminal fragment of an L-chain.
In 1838, Mathias Schleiden (a German botanist) coined the term amyloid to describe the normal amylaceous constituent of plants. In 1854, Rudolf Virchow used the term amyloid. Virchow described its reaction with iodine and sulfuric acid, which, at the time, was a marker for starch; thus, the term amyloid or starchlike is used. Virchow adopted the term to describe abnormal extracellular material that is seen in the liver during autopsy.
Some 70 years after Virchow's description, Divry and associates recognized that the amyloid deposits showed apple-green birefringence when specimens stained with Congo red were viewed under polarized light. This observation remains the sine qua non of the diagnosis of amyloidosis.2
In 1959, with the use of electron microscopy, Cohen and Calkins first recognized that all forms of amyloidosis demonstrated a nonbranching fibrillar structure. Electron microscopy remains the most sensitive method for recognizing the disorder.3
Pathophysiology
The final pathway in the development of amyloidosis is the production of amyloid fibrils in the extracellular matrix. The process by which precursor proteins produce fibrils appears to be multifactorial and differs among the various types of amyloidosis.
The fibrils in primary systemic amyloidosis are composed of Ig L-chain material (protein amyloid L) consisting of intact L-chains, L-chain fragments (particularly the variable amino-terminal region), or both. Amyloid deposition occurs as a result of plasma-cell dyscrasia.
The diagnosis depends on the demonstration of amyloid deposits in tissue. The organs most commonly involved are the kidneys or heart, either individually or together.4 Autonomic and sensory neuropathies are relatively common features.
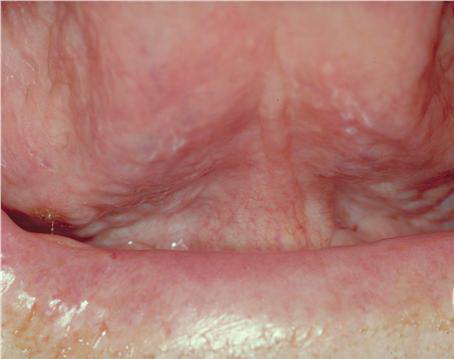
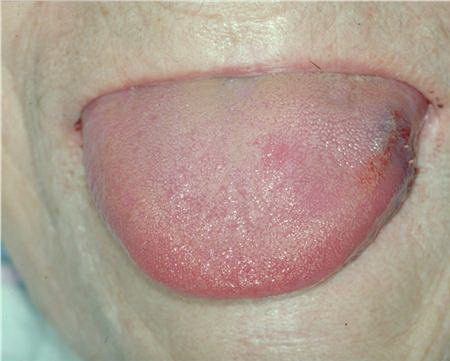
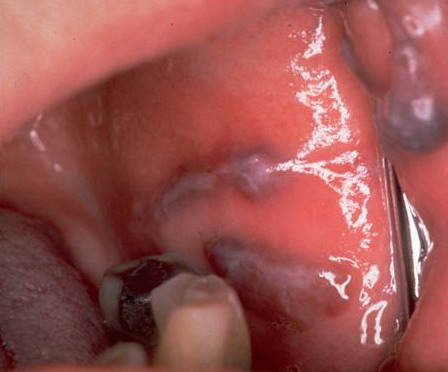
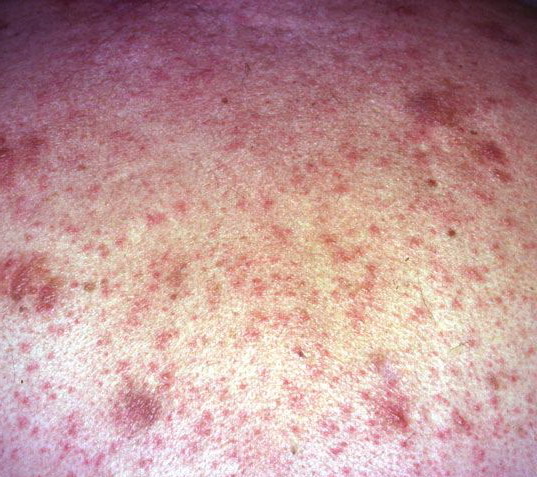
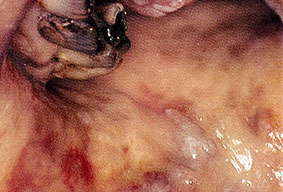
About 30-40% of patients with primary systemic amyloidosis have cutaneous findings. Mucocutaneous involvement provides early evidence of the existence of an underlying plasma-cell dyscrasia. Petechiae, purpura, and ecchymoses that occur spontaneously or after minor trauma are the most common skin signs and are found in about 15-20% of patients.5 The most characteristic skin lesions consist of papules, nodules, and plaques that are waxy, smooth, and shiny.6 Scalp involvement may be evident with hair loss. Mucocutaneous changes in the oral cavity include localized rubbery papules, petechiae, and ecchymoses. Xerostomia may result from the infiltration of the salivary glands. Macroglossia is reported in 19% of patients with primary systemic amyloidosis.
Primary systemic amyloidosis accounts for 7% of nonhematological malignancies,7 but few cases of gastric carcinoma in patients with primary amyloidosis have been described. Although acute pseudoobstruction is an uncommon clinical manifestation of amyloidosis, the coexistence of both gastrointestinal hemorrhage and pseudoobstruction of the small intestine should alert the clinician to a diagnosis of gastrointestinal amyloidosis
History
The symptoms of a patient with primary systemic amyloidosis (PSA) are rarely helpful in making the diagnosis because they are often too nonspecific. Therefore, the diagnosis is often delayed.
- Presenting symptoms include the following:
- Fatigue
- Weight loss
- Paresthesias
- Hoarseness
- Edema
- Classically, patients present with symptoms of the following:
- Carpal tunnel syndrome
- Macroglossia
- Mucocutaneous lesions
- Hepatomegaly
- Edema
- The organs most commonly involved are the kidneys or heart, either individually or together.
- The patients' symptoms reflect the organ or organs most prominently involved.
Physical
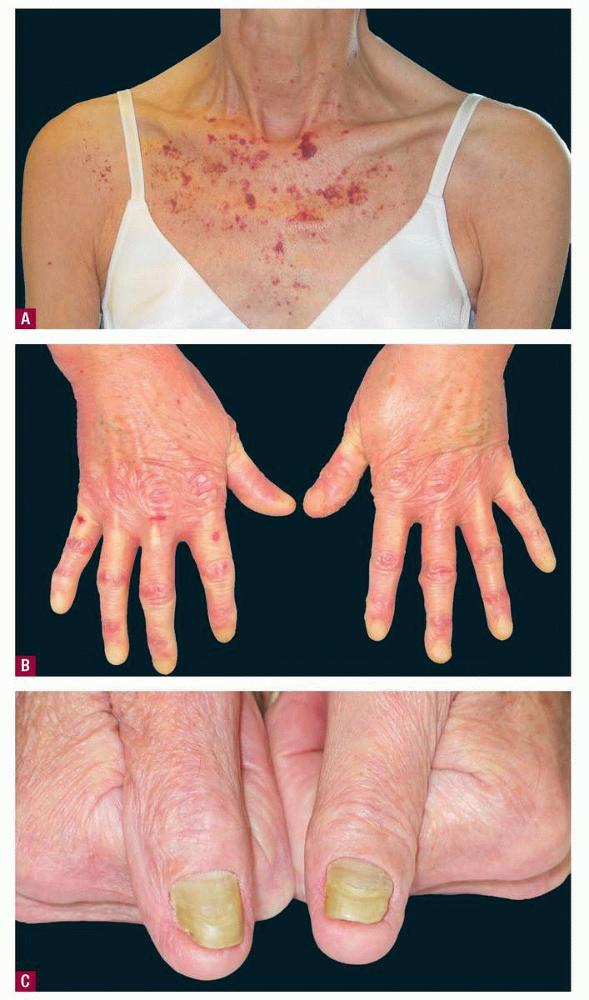
- Clinically evident mucocutaneous involvement occurs in 30-40% of patients with primary systemic amyloidosis, and it provides an early clue to the existence of an underlying plasma-cell dyscrasia.
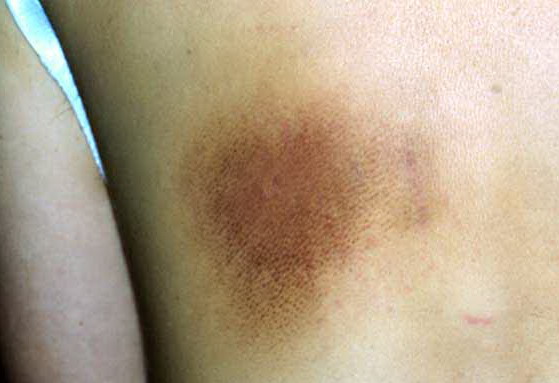
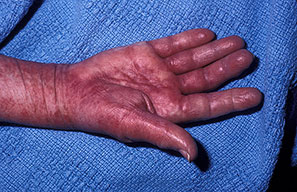
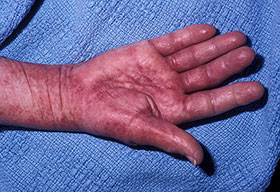
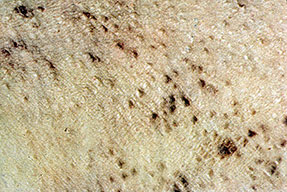
- Petechiae and ecchymoses are the most common skin findings, because of cutaneous blood vessel involvement.
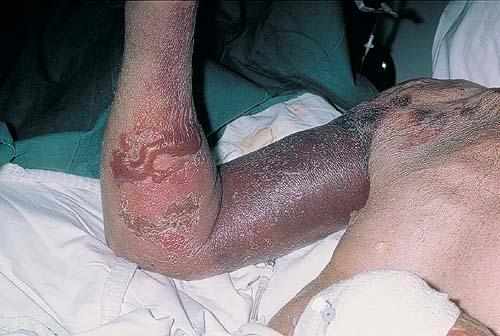
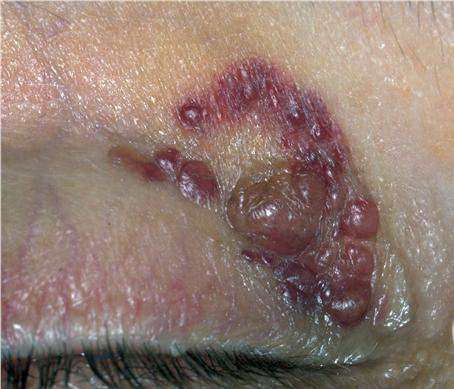
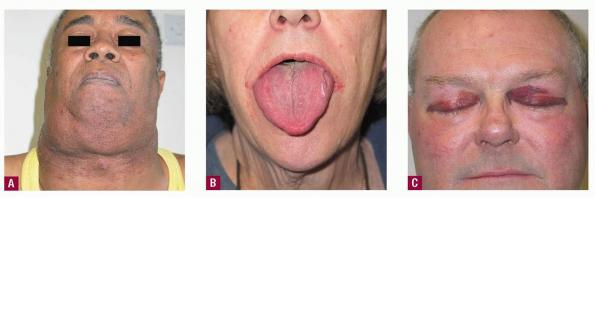
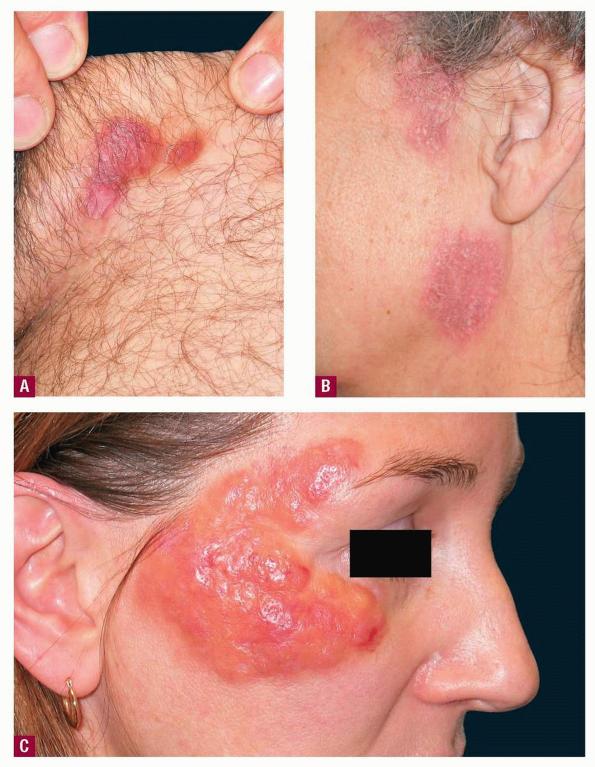
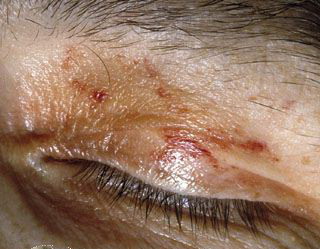
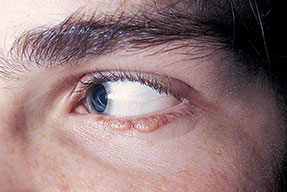
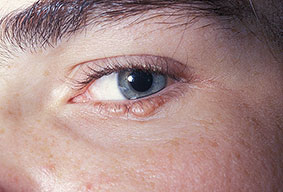
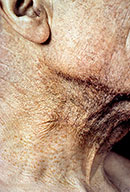
- The face is most commonly affected; minor trauma sometimes precipitates eyelid and periorbital purpura (pinch purpura or raccoon eyes sign). Purpuric lesions are found in flexural regions such as the nasolabial folds, neck, and axillae.
- At times, bullae form; these may be hemorrhagic.
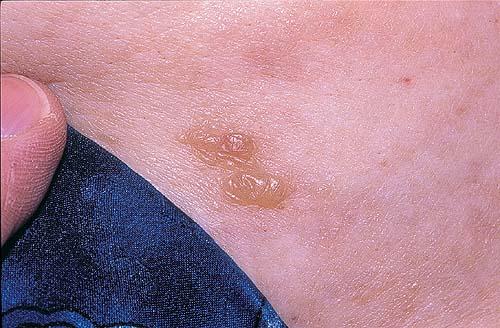
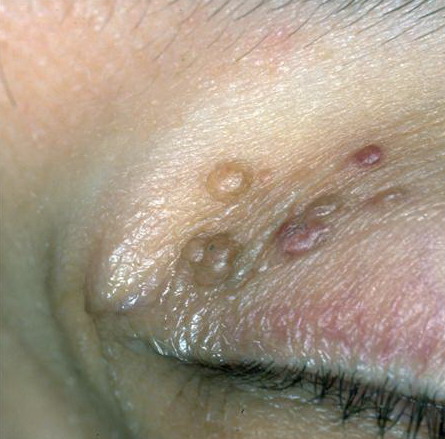
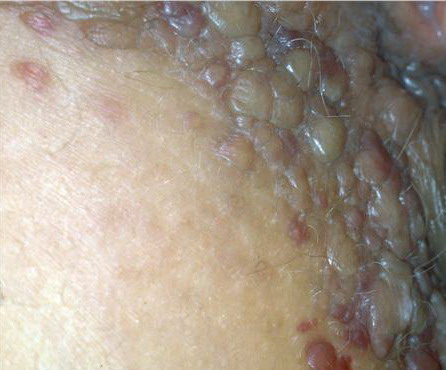
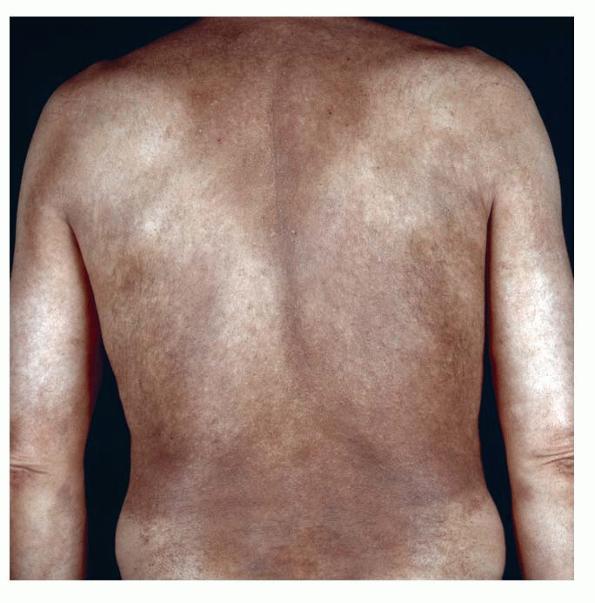
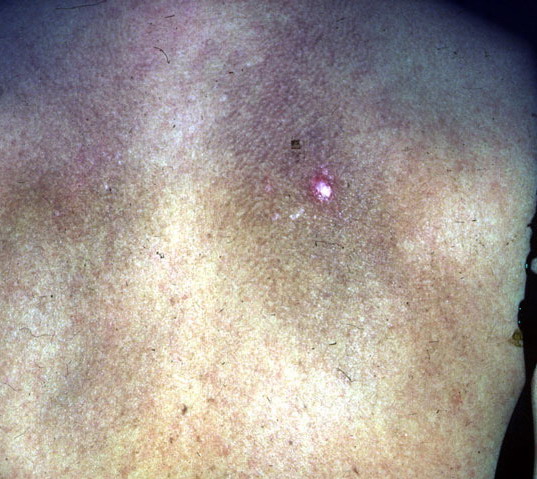
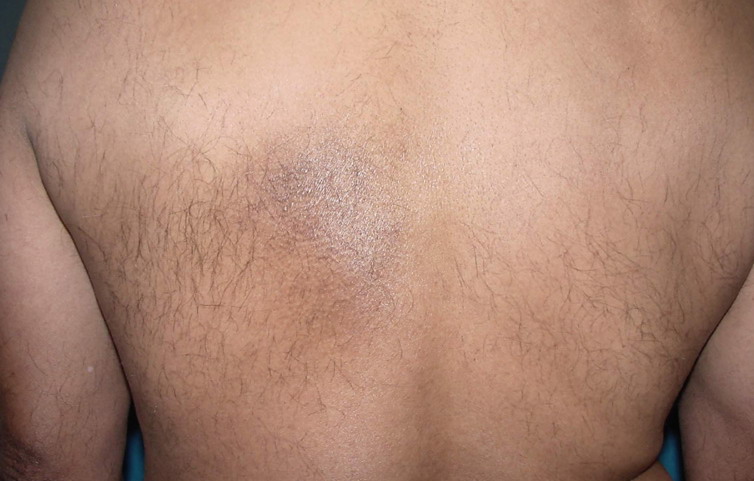
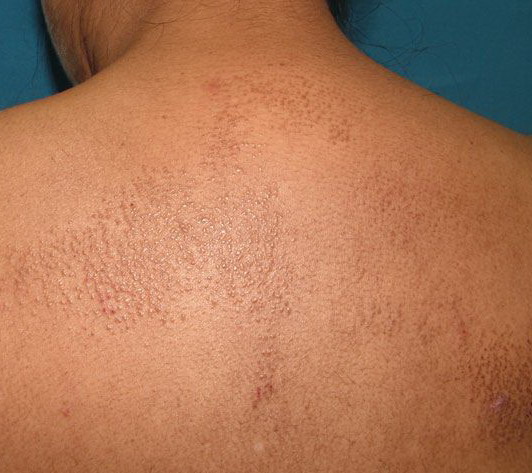
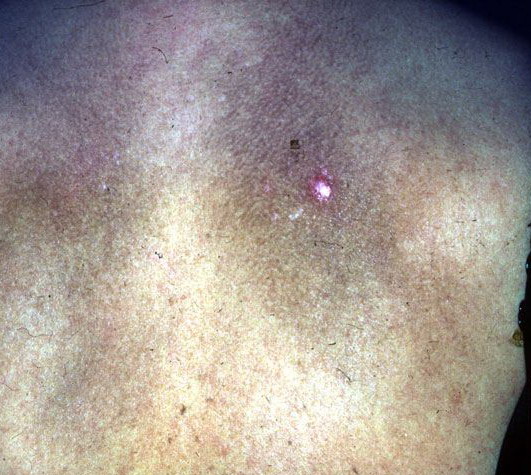
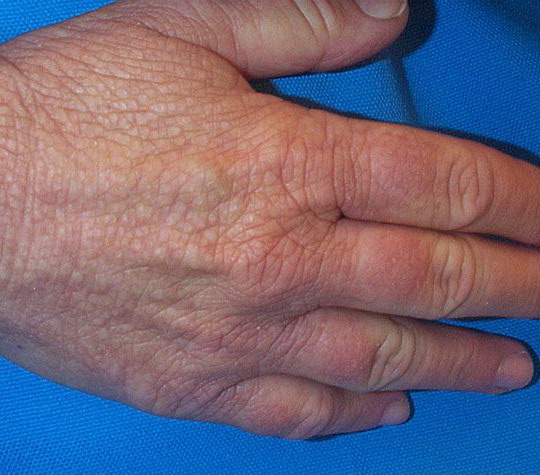
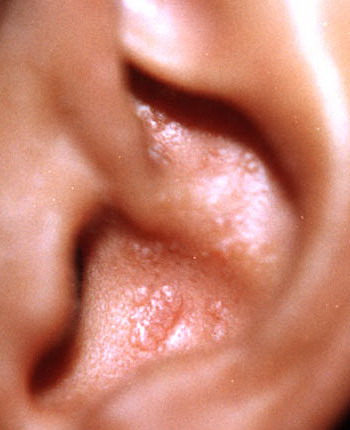
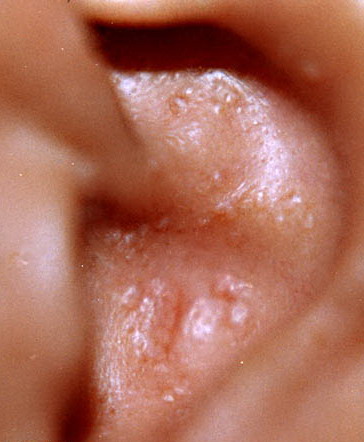
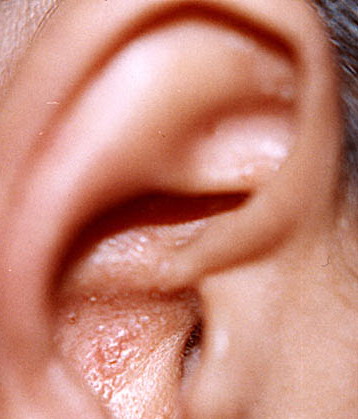
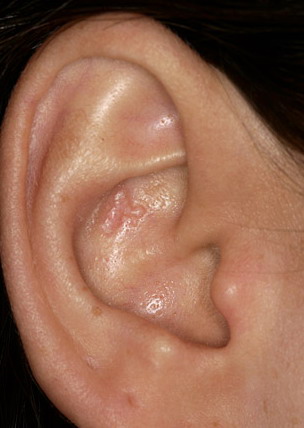
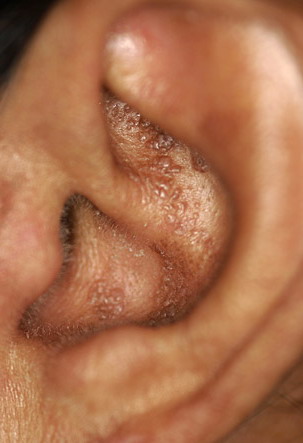
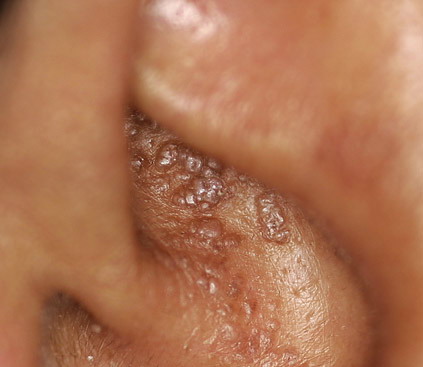
- The most characteristic skin lesion in primary systemic amyloidosis consists of waxy papules, nodules, or plaques that may be evident in the eyelids, retroauricular region, neck, or inguinal and anogenital regions. Plaques may coalesce to form large tumefactive lesions.
- Diffuse infiltrates may resemble infiltrates of scleroderma or myxedema.
- Scalp involvement may appear as diffuse or patchy alopecia.11
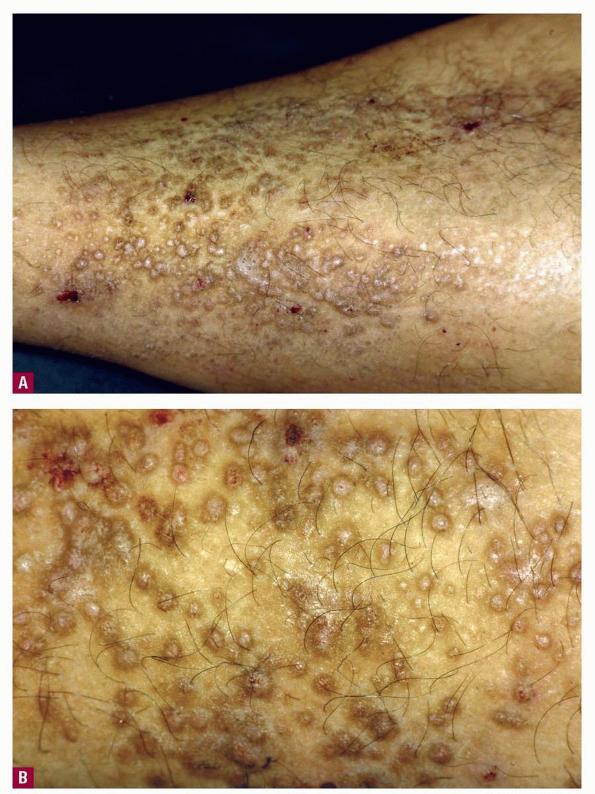
- Dystrophic nail changes include brittleness, crumbling, and subungual striation.
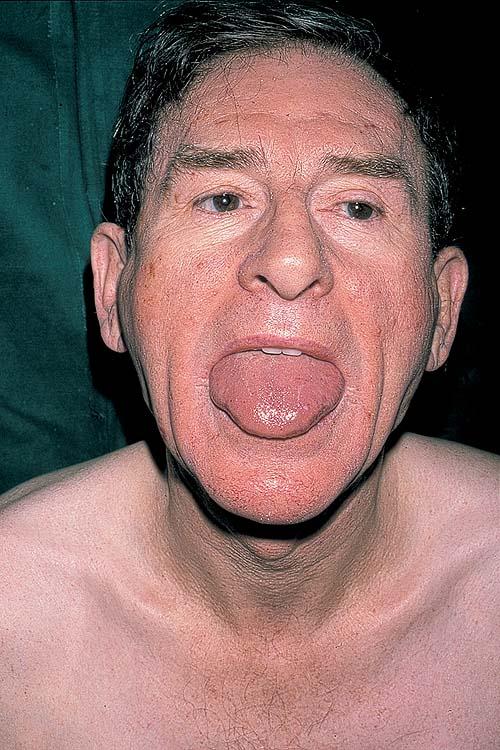
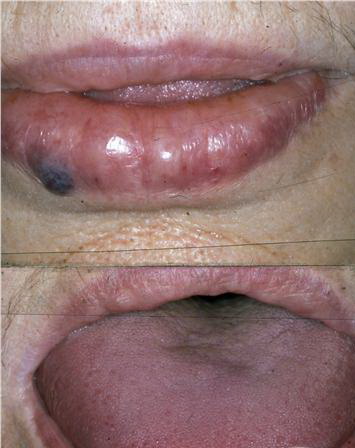
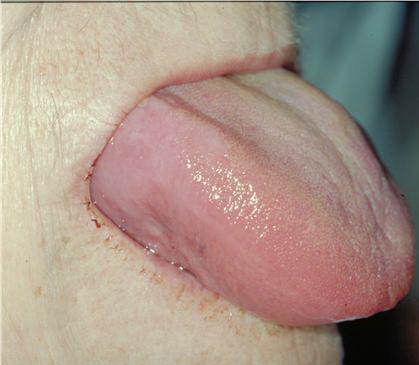
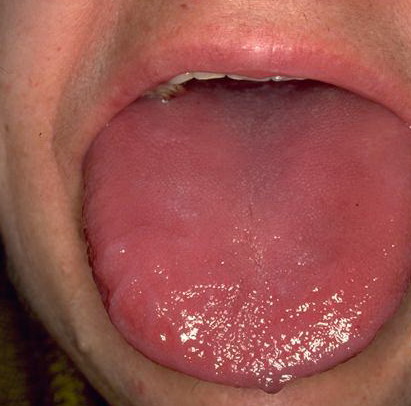
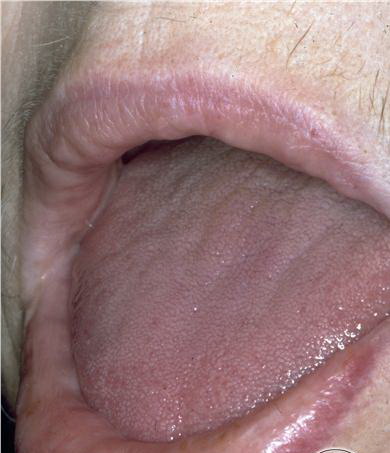
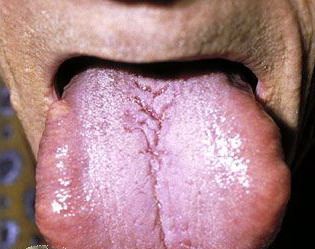
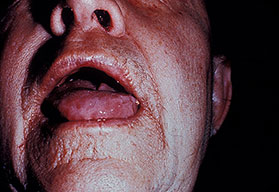
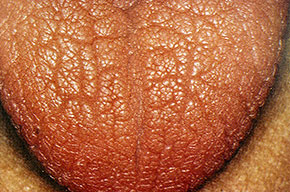
- The tongue may be infiltrated, resulting in macroglossia. Macroglossia is a classic feature of primary systemic amyloidosis. The tongue may extrude through gaps between the teeth to produce unique irregular indentations. The presence of amyloid in the oral cavity is often revealed by localized, soft, elastic papules.12,13
- Amyloid deposition in the smooth and striated muscles, connective tissue, blood vessel walls, and peripheral nerves may result in myocardial insufficiency, which is the most common cause of death in this fatal disease.
- Cardiac infiltration may cause angina, infarction, arrhythmias, or orthostatic hypotension.
- Blood vessel infiltration may lead to claudication of the legs or jaw.
- Renal amyloidosis usually manifests as proteinuria, often resulting in nephrotic syndrome.
- Edema is frequently found and may be the result of cardiac failure or nephrotic syndrome.
- Amyloid infiltration of the gastrointestinal tract may result in hemorrhage or malabsorption. Gut bleeding may also be fatal.
- Hepatomegaly occurs in about 50% of patients with primary systemic amyloidosis, but splenomegaly is present in less than 10% of patients.
- Autonomic and sensory neuropathies are relatively common features. Autonomic neuropathy may result in symptomatic postural hypotension, impotence, and disturbances in gastrointestinal motility.
- Summers and Kendrick reported and association with CREST syndrome (ie, calcinosis, Raynaud phenomenon, esophageal dysmotility, sclerodactyly, and telangiectasia syndrome).14
- Villa et al reported on amyloid goiter.15
Causes
Primary systemic amyloidosis is a plasma-cell dyscrasia characterized by an autonomous proliferation of plasma cells with an overproduction of a monoclonal Ig protein.
Laboratory Studies
- In a review of 132 primary systemic amyloidosis cases, Kyle and Bayrd reported that laboratory studies revealed anemia in less than 50% of the cases.8 The white cell count was usually within the reference range, and the erythrocyte sedimentation rate was higher than 50 mm/h in one half of the cases. Hepatic function was abnormal, and the serum creatinine level was increased in 50% of patients. Proteinuria was present in more than 90% of the cases.
- Conventional urine heat testing and electrophoresis of serum and urine samples may fail to demonstrate small quantities of monoclonal paraprotein or Bence-Jones protein. Immunoelectrophoresis of serum and concentrated urine samples is essential.
Imaging Studies
- Echocardiography is valuable in the evaluation of amyloid heart disease. It usually reveals a concentrically thickened left ventricle and often a thickened right ventricle, with a normal-to-small cavity.
- Doppler studies are useful and may show abnormal relaxation early in the course of the disease. Advanced involvement is characterized by restrictive hemodynamics.
Procedures
- Biopsy of a cutaneous lesion, if present, has the advantage of safety and a high diagnostic yield.
- Biopsy results in clinically normal skin may be positive in as many as 50% of cases of primary systemic amyloidosis.
- Findings from abdominal fat aspiration are positive in almost 80% of patients.
- Rectal biopsy reveals positive findings in about 80% of patients.
- If specimens from the biopsy sites are negative for amyloid, tissue should be obtained from an organ or area with suspected involvement, such as the kidney, liver, heart, or sural nerve.
Histologic Findings
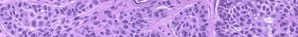
The best way to identify amyloid is to stain paraffin-embedded sections with alkaline Congo red and to examine them with polarized light to elicit a green fluorescence. Routine hematoxylin-eosin staining may show a homogenous, faintly eosinophilic mass if enough amyloid is present.
Analysis of a skin biopsy specimen of a papule reveals an amorphous or fissured eosinophilic mass in the papillary dermis with associated thinning or obliteration of the rete ridges. Nodules and plaques may demonstrate diffuse amyloid deposition in the reticular dermis or subcutis. Amyloid depositions are usually not associated with an inflammatory infiltrate.
The appearance of amyloid infiltration of the blood vessel walls, pilosebaceous units, arrector pili muscles, and lamina propria of sweat glands and infiltration around individual fat cells in the subcutis (known as amyloid rings) are characteristic findings. Amyloid may be deposited in the nail bed of dystrophic nails.
.
Medical Care
The treatment of primary systemic amyloidosis (PSA) is directed toward the affected organ and the specific type of the disease. In studies of different regimens of intermittent oral melphalan and prednisone, Skinner et al and Kyle et al reported that the response rates were low, with an increased survival from a median of approximately 7-9 months in patients who did not receive chemotherapy to approximately 12-18 months in those receiving chemotherapy.9,17
Shimojima et al reported a patient with primary systemic amyloidosis who achieved partial hematological response after 2 courses of the VAD (vincristine, doxorubicin [Adriamycin], and dexamethasone) chemotherapy regimen and subsequent high-dose melphalan followed by autologous peripheral blood stem cell transplantation despite involvement of multiple organs, including the heart.18 When amyloidosis-related dysfunction is seen in multiple organs, intensive chemotherapy might be a possible therapeutic option, although several modifications in the regimen and careful management are necessary.
- The nephrotic syndrome requires supportive therapy and diuretics, and renal failure can be successfully treated with dialysis.
- Congestive heart failure may respond to diuretics, but larger doses are often required as the disease progresses. The use of calcium channel blockers, beta-blockers, and digoxin are contraindicated in cardiac amyloidosis, because they may cause toxicity at therapeutic levels.
- Gastrointestinal involvement and neuropathy are treated symptomatically.
Medication
The treatment of primary systemic amyloidosis is often unsatisfactory. No reliable method for the accurate assessment of the total amount of amyloid in the body exists. Investigations are limited to the evaluation of organ function and the measurement of monoclonal protein levels in the serum and urine.
The similarity between primary systemic amyloidosis and multiple myeloma suggests that chemotherapy may be useful. Using different regimens of intermittent oral melphalan and prednisone, 2 groups of investigators9,17 confirmed the effectiveness of this therapy compared with no therapy or therapy with colchicine alone. However, the response rate was low, with an increased survival from a median of approximately 7-9 months in patients who did not receive chemotherapy to approximately 12-18 months in those receiving chemotherapy.
In another trial, Kyle and Greipp reported the effectiveness of combined melphalan and prednisone therapy compared with placebo therapy.19 Although the nephrotic syndrome improved in a number of individuals receiving the active medications, overall survival rates for the active and placebo groups were not substantially different.
Colchicine has also been used in the treatment of primary systemic amyloidosis. Colchicine may inhibit amyloid deposition by blocking the formation of amyloid-enhancing factors, and it also inhibits the secretion of amyloid from hepatocytes.
Based on encouraging results in myeloma patients, Dispenzieri et al reported results of a clinical trial of lenalidomide therapy with or without dexamethasone in patients with primary systemic amyloidosis.20 As a single agent, lenalidomide had modest activity in primary systemic amyloidosis. This activity was significantly enhanced when lenalidomide was used in conjunction with dexamethasone.
Immunosuppressants
These agents inhibit key factors in the immune system that are responsible for inflammatory responses.
Prednisone (Deltasone, Orasone, Meticorten)
Immunosuppressant for treatment of autoimmune disorders; may decrease inflammation by reversing increased capillary permeability and suppressing PMN activity. Stabilizes lysosomal membranes and suppresses lymphocytes and antibody production.
Adult
5-60 mg/d PO qd or divided bid/qid; taper over 2 wk as symptoms resolve
Pediatric
4-5 mg/m2/d PO; alternatively, 0.05-2 mg/kg PO divided bid/qid; taper over 2 wk as symptoms resolve
Coadministration with estrogens may decrease clearance; concurrent digoxin may cause digitalis toxicity secondary to hypokalemia; phenobarbital, phenytoin, and rifampin may increase metabolism of glucocorticoids (consider increasing maintenance dose); monitor for hypokalemia with coadministration of diuretics
Documented hypersensitivity; viral, fungal, tubercular, or connective tissue infection; peptic ulcer disease, hepatic dysfunction; GI disease
Pregnancy
B - Fetal risk not confirmed in studies in humans but has been shown in some studies in animals
Precautions
Abrupt discontinuation of glucocorticoids may cause adrenal crisis; hyperglycemia, edema, osteonecrosis, myopathy, peptic ulcer disease, hypokalemia, osteoporosis, euphoria, psychosis, myasthenia gravis, growth suppression, and infections may occur with glucocorticoid use
Dexamethasone (Decadron)
Has many pharmacologic benefits but significant adverse effects. Stabilizes cell and lysosomal membranes, increases surfactant synthesis, increases serum vitamin A concentration, and inhibits prostaglandin and proinflammatory cytokines (eg, TNF-alpha, IL-6, IL-2, and IFN-gamma). The inhibition of chemotactic factors and factors that increase capillary permeability inhibits recruitment of inflammatory cells into affected areas. Suppresses lymphocyte proliferation through direct cytolysis and inhibits mitosis. Breaks down granulocyte aggregates, and improves pulmonary microcirculation. Important chemotherapeutic agent in the treatment of ALL. Used in induction and reinduction therapy and given as intermittent pulses during continuation therapy.
Adverse effects are hyperglycemia, hypertension, weight loss, GI bleeding or perforation synthesis, cerebral palsy, adrenal suppression, and death. Most of the adverse effects of corticosteroids are dose or duration dependent.
Readily absorbed via the GI tract and metabolized in the liver. Inactive metabolites are excreted via the kidneys. Lacks salt-retaining property of hydrocortisone.
Patients can be switched from an IV to PO regimen in a 1:1 ratio.
Adult
6-8 mg/m2/d PO divided tid
Pediatric
Effects decrease with coadministration of barbiturates, phenytoin, and rifampin; decreases effect of salicylates and vaccines used for immunization
Documented hypersensitivity; active bacterial or fungal infection
Pregnancy
D - Fetal risk shown in humans; use only if benefits outweigh risk to fetus
Precautions
Increases risk of multiple complications, including severe infections; monitor adrenal insufficiency when tapering drug; abrupt discontinuation of glucocorticoids may cause adrenal crisis; hyperglycemia, edema, osteonecrosis, myopathy, peptic ulcer disease, hypokalemia, osteoporosis, euphoria, psychosis, myasthenia gravis, growth suppression, and infections are possible complications of glucocorticoid use
Antineoplastic agents
These agents inhibit cell growth and proliferation.
Melphalan (Alkeran)
Inhibits mitosis by cross-linking DNA strands.
Adult
0.15 mg/kg/d PO for 7 d or 0.25 mg/d for 4 d
Pediatric
4-20 mg/m2/d PO for 1-21 d
Concurrent administration with cyclosporine increases nephrotoxicity; cimetidine and H2 antagonists increase gastric pH decreasing effects; may exacerbate bone marrow suppression if administered 24 h before or 24 h after colony-stimulating factor; coadministration with zidovudine may contribute to bone marrow suppression
Documented hypersensitivity; severe bone marrow depression
Pregnancy
D - Fetal risk shown in humans; use only if benefits outweigh risk to fetus
Precautions
Amenorrhea may occur; caution in previously diagnosed myelosuppression
Uricosuric agents
These agents may inhibit the events involved in the inflammatory response associated with the disease.
Colchicine
Decreases leukocyte motility and phagocytosis in inflammatory responses.
Adult
0.5-1.2 mg PO initially, followed by 0.5-0.6 q1-2h or 1-1.2 mg q2h until a satisfactory response is attained; not to exceed 4 mg/d
1-3 mg IV initially, followed by 0.5 mg q6h until a satisfactory response is attained; not to exceed 4 mg/d
Pediatric
<12 years: Not established
>12 years: Administer as in adults
Significantly increases sympathomimetic agent toxicity and effect of CNS depressants
Documented hypersensitivity; severe renal, hepatic, GI, or cardiac disorders; blood dyscrasias
Pregnancy
D - Fetal risk shown in humans; use only if benefits outweigh risk to fetus
Precautions
Risk of renal failure, hepatic failure, permanent hair loss, bone marrow suppression, numbness or tingling in hands and feet, disseminated intravascular coagulopathy, and decreased sperm count; dose-dependent GI upset and diarrhea common
Immunomodulators
Lenalidomide (Revlimid)
Structurally similar to thalidomide. Elicits immunomodulatory and antiangiogenic properties. Inhibits proinflammatory cytokine secretion and increases anti-inflammatory cytokines from peripheral blood mononuclear cells.
Adult
10 mg PO qd initially; dose adjustment required if renal impairment, thrombocytopenia, or neutropenia occurs
Pediatric
<18 years: Not established
>18 years: Administer as in adults