FOCAL DERMAL HYPOPLASIA
OF GOLTZ
Epidemiology
Focal dermal hypoplasia of Goltz (FDH), first described in 1934, received its name from a report by Goltz and co-authors of three patients. Subsequently, there have been over 200 case reports published.
Etiology, Pathogenesis, and Genetics
Focal dermal hypoplasia is an X-linked dominant disorder, usually lethal in
males. Case reports of males with the condition are believed to be due to mosaicism for postzygotic mutations (as is true for incontinentia pigmenti); the presence of some normal cells allows survival in the male. The mutated gene is PORCN, the human homologue of the porcupine gene in drosophila. PORCN is thought to be important for palmitoylation and secretion of Wnt protein, a key regulator of the development of skin and bone.
Clinical Manifestations
DERMATOLOGIC
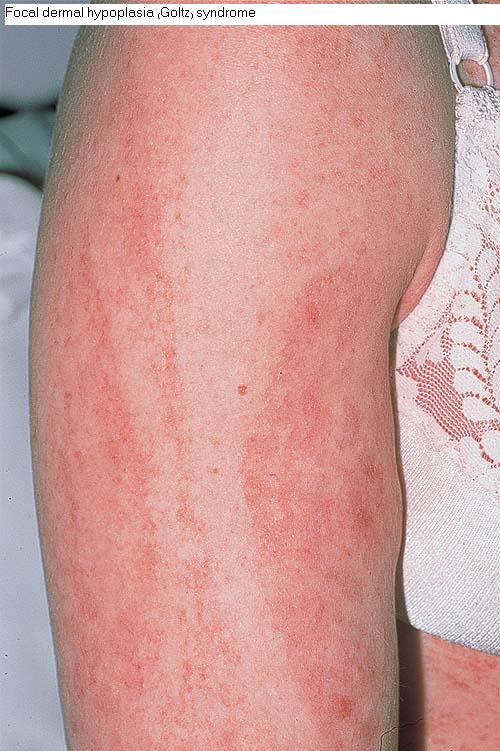
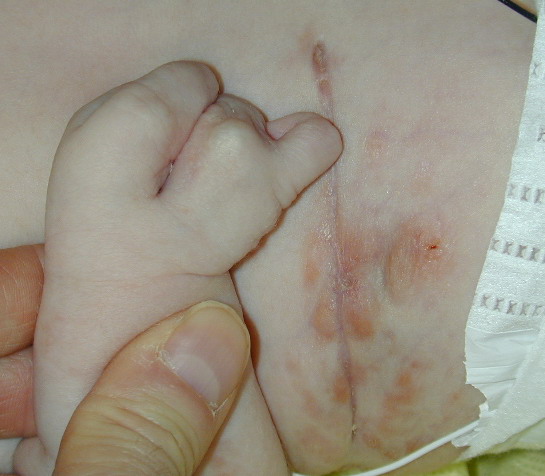
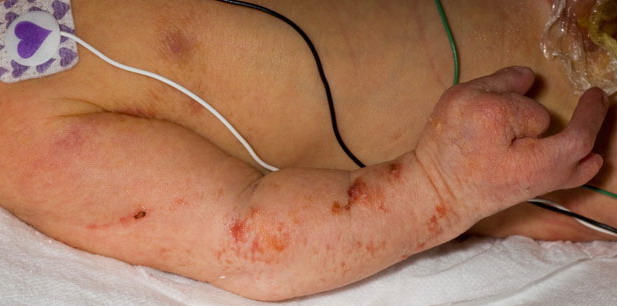
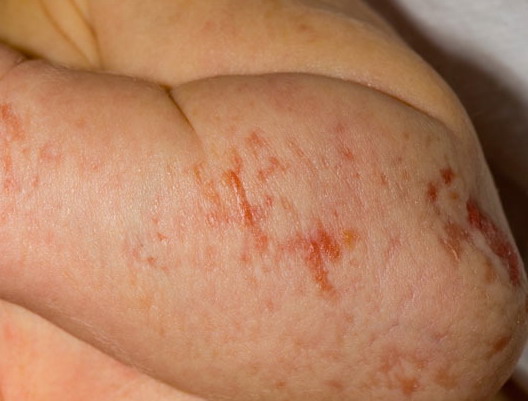
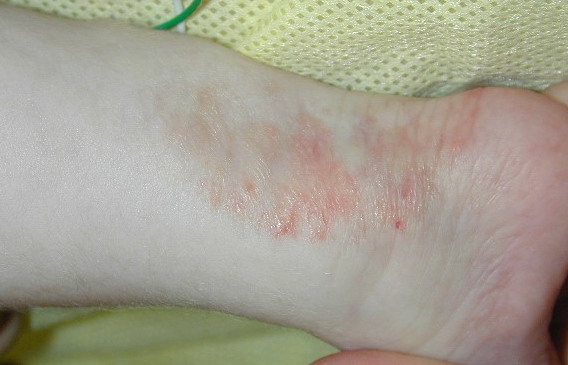
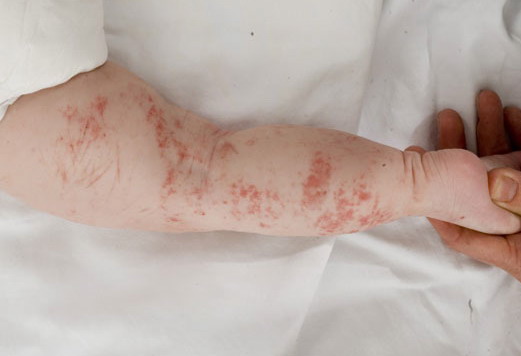
The skin changes of FDH are the primary diagnostic features. There is linear, punctate, streaky cribriform atrophy with telangiectasia. The cribriform atrophy is marked by tiny ice pick-like depressions in the skin. These are distributed along the lines of Blaschko. Areas of thinned to absent dermis are irregularly distributed and the resultant herniations of fat appear as yellow-pink excrescences on the skin surface . These are easily depressed. Papillomas that may be fleshy or vascular develop throughout life and favor the perigenital, perioral, intertriginous, and mucosal surfaces.
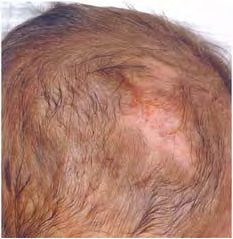
Other dermatologic features include patchy alopecia, brittle or sparse hair, and palmar and plantar hyperkeratoses. Some individuals have had hyperhidrosis and some have had aplasia cutis congenita.
SYSTEMIC ASSOCIATIONS
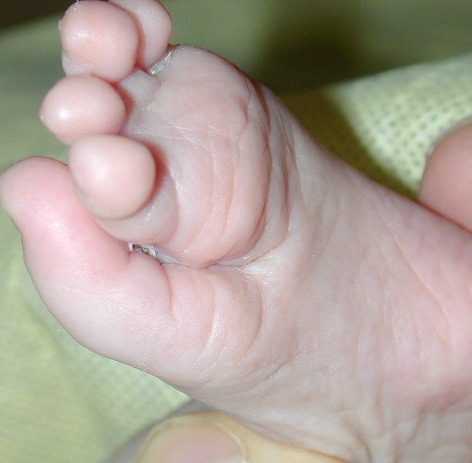
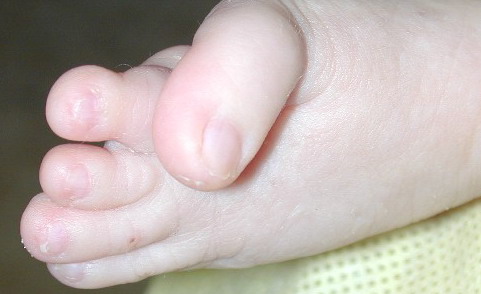
The other organ systems most frequently involved in FDH are the skeletal, central nervous system, teeth, and eyes. Microphthalmia and coloboma are common and the diagnosis of FDH should prompt a full ophthalmologic evaluation. Oligodontia, tooth dysplasia, and enamel defects are common. The skeletal abnormalities are too numerous to list; the more common are vertical banding of the bones (osteopathia striata), syndactyly (both cutaneous and bony), asymmetry, and short stature. Mental retardation has been reported in approximately 15 percent of cases.
Defects in other organ systems have been described in a minority of cases, including cardiac defects, abdominal wall defects, and renal malformations. It is not certain that these are inherent features of FDH.
Diagnosis and Differential Diagnosis
The diagnosis of FDH is relatively straightforward. Cribriform atrophy has been described in X-linked dominant Conradi-Hünermann syndrome (chondrodysplasia punctata), but ichthyosis is not a feature of FDH, and fat herniation is not part of Conradi-Hünermann. The streaky distribution of the atrophic lesions of IP is similar, as are the other system malformations, but the blistering, hyperkeratosis, and hyperpigmentation of IP are not found in FDH. In microphthalmia and linear skin defects/microphthalmia, dermal aplasia, sclerocornea, the skin defects are limited to the head and neck; there is atrophy and scarring of the skin more similar to aplasia cutis congenita and not dermal atrophy alone. The disorders do share similar ocular abnormalities.
Treatment and Prognosis
There is no specific treatment for the dermatologic and systemic features of FDH. Papillomas can be excised if they interfere with function. Use of vascular lasers to decrease the erythema of telangiectatic areas may have cosmetic benefit. As with most X-linked dominant disorders, clinical involvement varies considerably, and the range in severity is marked. This makes prognostic counseling difficult early in infancy, and usually it is wiser to counsel patience and reassurance until the extent to which there is systemic involvement becomes clear. It is important to inquire about the family history of lost pregnancies (a distorted male-female ratio in offspring and increased pregnancy loss are clues to the mother being a carrier). Both mothers and fathers should be examined carefully; fathers may have subtle features, and, presumably, represent individuals with postzygotic mutations for the FDH gene.