Erythropoietic protoporphyria
Erythropoietic protoporphyria is a genetic disorder most often arising from impaired activity of ferrochelatase, the ultimate enzyme of heme biosynthesis.1,2 The resultant accumulated excess of its substrate, protoporphyrin, causes 2 principal manifestations: (1) a distinctive acute cutaneous photosensitivity typically appearing in childhood and (2) hepatobiliary disease.1,3,4,5
The predominant genotype associated with phenotypic expression is one mutant ferrochelatase allele encoding a defective enzyme protein with little or no function, coupled with a normal variant allele with low gene expression.6,7 Infrequently, 2 deleterious mutations are found in symptomatic individuals; this recessively inherited form of protoporphyria appears to impart a higher risk for hepatic dysfunction.8,9
A recently described X-linked dominant form of protoporphyria arises from C-terminal deletions in the gene encoding the erythroid-specific enzyme 5-aminolevulinic acid synthase-2; increased function of this enzyme leads to overproduction of protoporphyrin with associated acute photosensitivity and increased risk for liver disease.10
Rarely, acquired somatic mutation or deletion of a ferrochelatase gene secondary to myelodysplastic or myeloproliferative disorders leads to an adult-onset protoporphyric disorder.11,12,13
Pathophysiology
Protoporphyrin is a lipophilic molecule capable of transformation to excited states by absorption of light energy. Excited-state protoporphyrin mediates photoxidative damage to biomolecular targets in the skin,14 resulting in immediate phototoxic symptoms variously described as tingling, stinging, or burning that may be followed by the appearance of erythema, edema, and purpura.3,14 Excess protoporphyrin is formed during maturation of erythroid cells in the bone marrow and is present at the highest levels in reticulocytes and young erythrocytes.15 Protoporphyrin escapes from red blood cells into the plasma, from which it is cleared by the liver and secreted into bile. Protoporphyrin-rich bile facilitates gallstone formation.16 Toxic effects of protoporphyrin deposition in the liver may lead to life-threatening hepatic dysfunction.
History
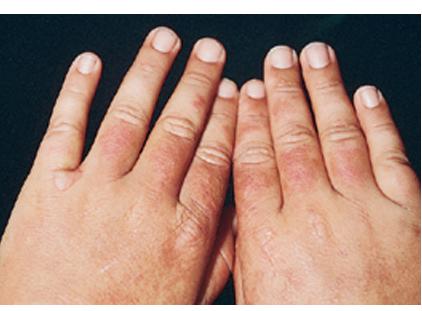
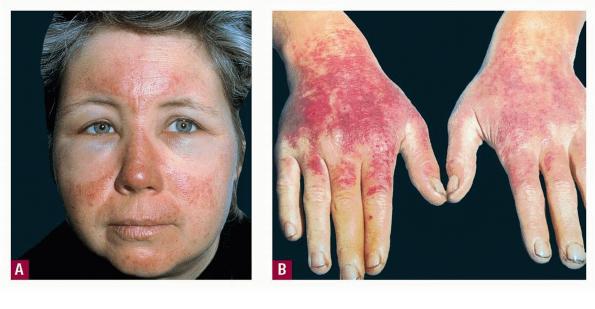
Uncomfortable sensations in skin exposed to sunlight typically begin during infancy or childhood, most often involving dorsal hands, the face and ears, and, occasionally, legs and dorsal feet, after short periods of exposure. If exposure is promptly discontinued, visible skin lesions may not ensue. Longer exposure, or multiple exposures on sequential days, can elicit swelling with or without redness in the exposed skin that evolves into sheets of petechiae. This exquisitely painful reaction resolves over several days to leave skin that may appear normal. Eventually, chronic changes may develop that are highly suggestive, but, when subtle, can be overlooked.
Individuals with protoporphyria who report skin pain but have minimal objective findings may be considered malingerers until an acute reaction is observed. Gallstones may remain silent or evoke reports of indigestion and/or right upper quadrant abdominal pain consistent with symptomatic cholelithiasis. Individuals with protoporphyria associated with hepatotoxicity may report loss of appetite, nausea, vomiting, weakness and fatigue, anorexia, malaise, weight changes, increasing abdominal girth, pain in the epigastrium or right upper quadrant and back, jaundice, and increasing photosensitivity.
Physical
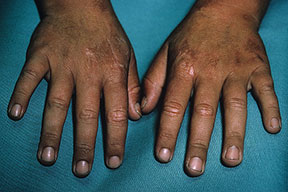
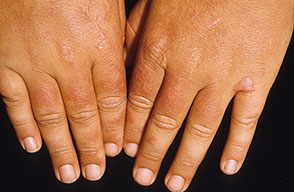
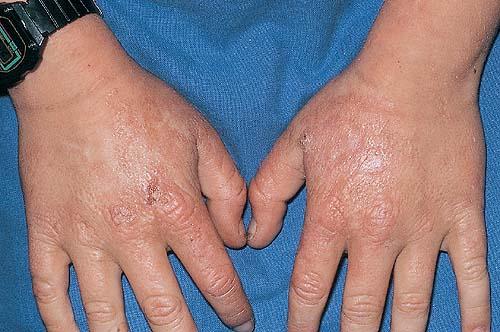
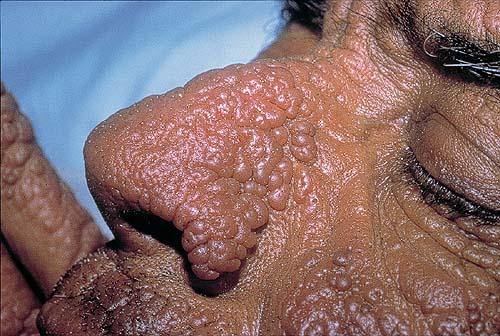
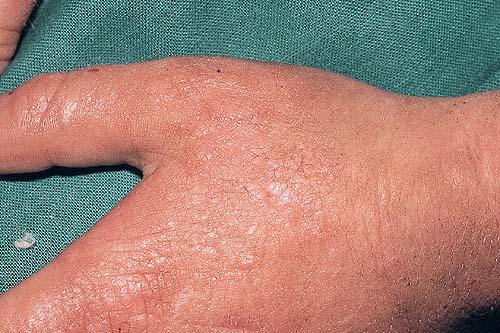
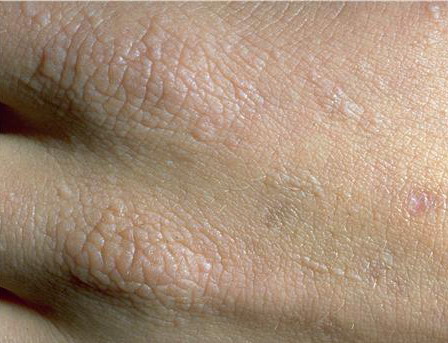
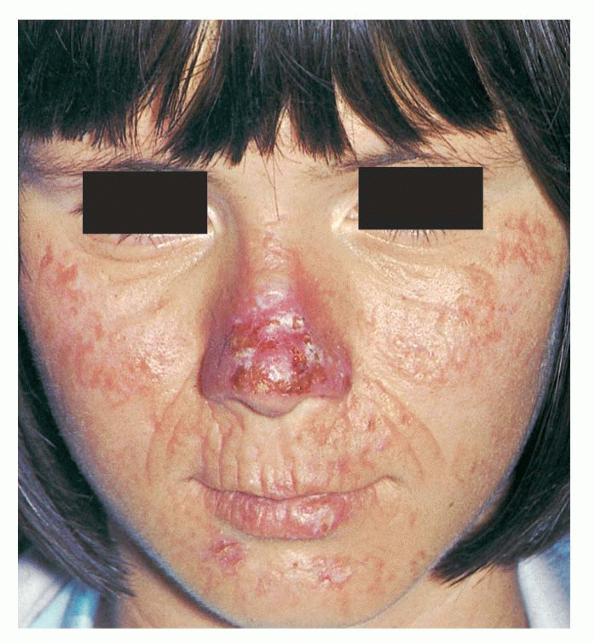
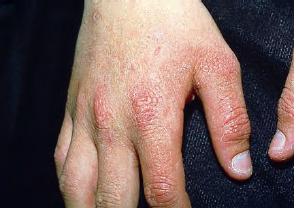
The acute phototoxic reaction typically includes edema, erythema, and petechiae. Blisters, crusted erosions, and scarring may occur but are less florid and less frequent than in other porphyrias. Chronic changes include shallow, elongated depressions in facial skin, especially over the nose; perioral furrowing; and prematurely aged, thickened, or coarsely textured skin of the dorsal hands, often most prominent over the knuckles. In more severe cases, sclerodermalike waxy induration or a cobblestone texture of the face and hands develops. Mechanical fragility, when present, is less severe than in other porphyrias; hypertrichosis is infrequent.
With progressive liver dysfunction, hepatosplenomegaly and jaundice may develop, as may signs of increasing cutaneous photosensitivity. End-stage liver disease is signaled by intense jaundice, ascites, vomiting, fever, encephalopathy, axonal polyneuropathy that may progress to paresis and respiratory failure, hemorrhage from esophageal varices, and extreme photosensitivity.
Causes
The ferrochelatase gene is located on band 18q21.3.23 Ferrochelatase mutations listed at the Human Gene Mutation Database numbered 124 as of May 2010.
Loss of ferrochelatase activity by as much as 50% as the result of 1 mutant gene is generally insufficient to cause overt disease when its complementary allele has normal function.6 Ferrochelatase genotypes composed of either 2 mutant alleles (approximately 4% of cases) or 1 mutation and a nonmutant allele with a specific intronic single nucleotide polymorphism (IVS3-48C) (approximately 94% of cases) have been found in most symptomatic individuals.8,9,24 This polymorphism enhances aberrant splicing and rapid degradation of ferrochelatase mRNA, with resultant low expression.7 The allele frequency of this polymorphism varies widely in diverse populations studied, as follows:
- Japanese - 43.3%9
- Southeast Asian - 31%9
- White French - 11.3%9
- North African - 2.7%9
- Black West African - <1%9
- United States - 3.5%25
- South Africans of European descent - 8.6%2
- United Kingdom - 6.5%8
- Chinese (3 different regions) - 28-41.4%26
- Swedish - 8%27
The pairing of a mutated allele encoding a severely impaired enzyme protein with this low-expressing polymorphic allele typically yields enzyme activity diminished to less than 30% of normal, low enough to cause protoporphyrin accumulation. Individuals heteroallelic or homoallelic for this polymorphism do not have sufficiently diminished ferrochelatase activity to cause clinical abnormalities, although their erythrocyte protoporphyrin levels may be mildly abnormal.2
Adult-onset protoporphyric photosensitivity and increased protoporphyrin levels have been associated with an acquired somatic mutation or deletion of a ferrochelatase gene due to myelodysplastic or myeloproliferative disorders.11,12,13
Eight families have been described recently with a protoporphyric disorder indistinguishable clinically from the predominant form of the disease, but without ferrochelatase mutations.10 Two different C-terminal deletions in the gene encoding the erythroid-specific isoform of aminolevulinic acid synthase were identified among these families. The locus for this gene is on the X chromosome, and the inheritance pattern in the families was consistent with X-linked dominant transmission. Both mutations caused a marked increase in activity of aminolevulinic acid synthase 2 that eventuated in large accumulations of erythrocyte-free protoporphyrin and zinc-protoporphyrin. Seventeen percent of affected individuals in that study exhibited overt liver disease, a significantly greater number than the 2-5% of individuals with ferrochelatase-deficient protoporphyria who develop this complication.
Laboratory Studies
Protoporphyrin concentration is elevated in red blood cells, plasma, bile, and feces. The diagnosis is usually made by finding the abnormal levels in erythrocytes and plasma. Urinary porphyrin levels are normal in patients without liver dysfunction. Abnormal coproporphyrinuria develops when liver function is deteriorating.28,29
Erythrocyte and plasma protoporphyrin levels are increased several-fold over the reference range. Fecal protoporphyrin excretion may be increased, but, in many patients, it remains within the reference range. In impending liver failure, the dynamic equilibrium between rates of protoporphyrin production and excretion is altered, producing progressively rising erythrocyte and plasma porphyrin levels and progressively diminishing fecal porphyrin excretion.28,30
Obtain a complete blood cell count and serum liver function panel at diagnosis. Monitor serum indices of liver function at 6- to 12-month intervals if baseline values are normal. If liver function is abnormal, complicating factors (eg, gallstones, viral hepatitis, alcohol or drug abuse, other toxic, infectious, immunologic, or metabolic storage disorders) should be excluded by appropriate testing.
Perform a hematological assessment of anemia. Individuals with protoporphyria often have mildly lowered hemoglobin and hematocrit levels, which do not cause symptoms and do not require treatment.3,31 The mean corpuscular volume may be below the normal limit.
Imaging Studies
If cholelithiasis is suspected, abdominal ultrasonography or other imaging procedures are indicated.
Other Tests
Impending liver failure may be signaled by progressively rising levels of urinary coproporphyrin.29 Urinary porphyrin levels are within normal limits in persons with uncomplicated erythropoietic protoporphyria. Protoporphyrin, being lipophilic, is not excreted by renal mechanisms and does not normally appear in urine. Coproporphyrin, which accumulates as a result of liver disease, has intermediate water solubility, and levels become abnormally elevated in the urine of patients developing protoporphyrin-induced hepatotoxicity.28
Measurement of ferrochelatase enzyme activity remains a research procedure. Mutation analysis of the ferrochelatase gene (ie, DNA testing) is performed at several porphyria research units in various countries and is now commercially available in the United States. See the American Porphyria Foundation for further information.
Procedures
In the event of overt liver dysfunction, liver biopsy is indicated. Some experts suggest that individuals with genotypes associated with higher risk of liver disease, such as a "null-allele" ferrochelatase mutation that encodes an enzyme with essentially no residual activity, mutations of both ferrochelatase alleles, one of the aminolevulinic acid 2 increased-function mutations, or with a family history of protoporphyric liver disease, may warrant liver biopsy even before liver function tests become abnormal.18,19 The presence of other risk factors for liver disease, such as viral hepatitis, hemochromatosis, or alcoholic or nonalcoholic fatty liver, increases the weight of argument for earlier liver biopsy.
Liver transplantation may be life saving, but it does not cure protoporphyria because the source of most of the excess protoporphyrin is the bone marrow. Continued overproduction of protoporphyrin eventually leads to protoporphyrin deposition in the engrafted liver, which may again become dysfunctional.22 While bone marrow transplantation is potentially curative, its risks have warranted its application in only a few cases to date.12,21,32 Research in animal models has shown promising developments in gene therapy strategies that may eventually be transferrable to humans.
Histologic Findings
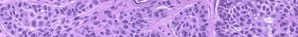
Light microscopy examination of the acute skin reaction shows perivascular and interstitial neutrophilic dermal infiltrates. Ultrastructural findings in the acute reaction include damage of endothelial cells with extravasation of intravascular contents and degranulated mast cells.33
Biopsy specimens of chronically damaged skin show deposition of hyaline masses in the upper dermis and markedly thickened walls of upper dermal capillaries.34 Ultrastructural findings in chronically damaged skin include replicated basal laminae around dermal vessels, degranulated mast cells, and amorphous dermal deposits.34 Direct immunofluorescence studies show deposition of immunoglobulins and complement in and around upper dermal vessel walls and, to a lesser extent, at the dermoepidermal junction.34
Liver biopsy typically reveals brown pigment in hepatocytes, Kupffer cells, portal macrophages, and small biliary structures.16,22 Many of these protoporphyrin deposits are crystalline when examined under electron microscopy and birefringent when examined under polarization microscopy.16,22 Cirrhotic changes are seen in advanced disease, including fibrous expansion of portal areas and regenerative nodules
Medical Care
For protoporphyria uncomplicated by hepatobiliary disease, the major problem is lifelong cutaneous photosensitivity. Anemia, if present, typically is mild and rarely requires specific therapy. Cholelithiasis is managed surgically. Liver dysfunction is an ominous development for which medical remedies are not consistently effective. Progressive intractable liver insufficiency is an indication for liver transplantation.16,18,35
Note the following treatment measures for photosensitivity:
- Shield skin from sunlight by using protective clothing and lifestyle adjustments.
- Because the wavelengths of light causing porphyrin-sensitized phototoxicity are chiefly in the visible spectrum, window glass is not an effective barrier. Plastic films that attenuate transmission of portions of the visible light and long UV spectra are available and can be applied to window or windshield glass.36
- Topical sunscreens are not effective unless transmission of long UV and visible light rays is reduced by their use. Sun-blocking formulations containing zinc oxide or titanium dioxide reflect visible light and may be helpful.36
- Topical sunless tanning gels or creams containing dihydroxyacetone produce superficial pigmentation that blocks some of the offending wavelengths.36
- Induction of endogenous melanin by exposure of skin to broad- or narrow-band UV-B lamps or to UV-A in conjunction with a psoralen UV-A photosensitizer also may increase tolerance to natural sunlight.37
- Afamelanotide, an alpha-melanocyte–stimulating hormone analogue that increases melanin production in the skin, is a novel injectable photoprotective agent currently in clinical trials in Australia and several European countries. It has recently become available by prescription in Italy.
- Oral beta-carotene reduces photosensitivity in some, but not all, patients.3,38,39
- Attenuation of photosensitivity using oral cysteine40 or pyridoxine41 has been reported but not widely confirmed.
- H1-receptor antagonists can mitigate histamine-mediated components of the acute reaction, but they rarely suppress all signs and symptoms.42 Suppression of heme synthesis by inhibition of cytochrome P-450 formation and of heme oxygenase activity is a mechanism proposed for transient improvement of isolated cases of various porphyrias after H2-receptor antagonist use that remains unproven.43
Although adverse reactions to porphyrinogenic drugs known to exacerbate acute hepatic porphyrias are not characteristic of protoporphyria, avoid or administer with caution drugs with cholestatic properties, such as estrogenic hormones. Assess the risk-to-benefit ratio for each individual with protoporphyria when considering use of cholestatic therapies.
Immunization against viral hepatitis agents should be offered.
Medical approaches to reversing protoporphyric liver dysfunction are not well established, owing to inconsistent or uncertain efficacy and experience in relatively few cases. Note the following:
- Orally administered cationic exchange resins or activated charcoal aimed at reducing enterohepatic recirculation of porphyrin and/or bile acids to enhance hepatic porphyrin excretion may have some level of efficacy in selected patients.28,44,45
- Hypertransfusion to slow erythropoiesis46 and intravenous infusion of heme analogues to repress endogenous porphyrin production47,48 have been beneficial in some cases.
- Administration of iron with the rationale of enhancing protoporphyrin conversion to heme appeared beneficial in one case,49 but aggravated the disease in others.50
- Reduction in erythrocyte protoporphyrin levels and improved liver function followed administration of vitamin E to a protoporphyric patient with cirrhosis.51
- Plasmapheresis48 or exchange transfusion52 to reduce the circulating protoporphyrin burden appeared helpful in a small number of advanced cases.
- Medical regimens are often used in combination or rapid sequence in progressively deteriorating patients and are best instituted by experts in a referral center for advanced liver disease.
Surgical Care
Surgical removal of gallstones usually poses no more risk for individuals with protoporphyria than for the general population, although phototoxic sequelae from high-intensity operating room lighting is a theoretical possibility. Adverse reactions to anesthetic agents problematic in acute hepatic porphyrias are not characteristic of protoporphyria. Failure of medical reversal of protoporphyrin-induced hepatic decompensation warrants liver transplantation. Operating room lamps have caused acute phototoxic damage to skin and internal organs during transplantation.53,54 Preoperative exchange transfusions, plasmapheresis, and/or infusion of a heme analogue may lower the circulating burden of protoporphyrin in the blood, reducing intraoperative phototoxic potential.55 These treatments may also aid postoperatively in retarding the development of protoporphyrin hepatotoxicity in the engrafted liver.47,48
Consultations
Consultation with a hematologist should be sought for management of anemia or if hypertransfusion, exchange transfusion, or plasmapheresis is considered. Rarely, bone marrow transplantation may have a role in the management of selected patients with severe manifestations.21,32
Referral to specialists at a comprehensive liver center should be arranged at the earliest signs of liver decompensation for assistance in evaluation and management of progressive liver dysfunction. If liver transplantation becomes necessary, a successful outcome is more likely if the procedure is performed before the patient is gravely debilitated.
Referral to a medical geneticist can aid in counseling patients and families about risks of inheriting or transmitting the mutations and polymorphisms associated with the disease.56,57
Preoperative consultation with anesthesiologists and biomedical engineers concerning operating room lighting is essential. The intense visible light emitted by surgical lamps can cause intraoperative burns of the skin and internal organs due to the massive protoporphyrin tissue accumulations that result from failure of hepatic excretory mechanisms.53,54 Filtering operating room lamps appropriately can block the most harmful portions of the visible light spectrum.55
Diet
Do not severely curtail carbohydrate intake; a beneficial glucose effect may be modulating abnormal heme synthesis.58 Limit use of ethanol; alcohol excess has been implicated in fatal protoporphyria associated with liver failure.59
Activity
Sunlight avoidance is mandatory. Recommend adjustment of outdoor activities to avoid midday sunlight. Stylish and comfortable sun-protective clothing is commercially available that can reduce time constraints on many outdoor sports or activities. Specialized programs for photosensitive children can be found that offer safe and healthy recreational experiences, even a summer camp organized by the Xeroderma Pigmentosum Society. See Camp Sundown.
Medication
The only oral photoprotective agent approved by the US Food and Drug Administration and widely used for the treatment of protoporphyria is a synthetic beta-carotene formulation now available over the counter as Lumitene. Cysteine has shown benefit in clinical trials. Pyridoxine was reported effective in 2 cases. H1-receptor blockade may reduce symptoms due to mast cell histamine release during acute phototoxic reactions if established prior to exposure. Whether H2-receptor antagonists reproducibly slow porphyrin production in various porphyrias remains unproven.
Liver dysfunction warrants individualized design of therapeutic regimens that may include the administration of enteric sorbents to promote protoporphyrin excretion, bile acids to enhance porphyrin clearance from the liver, and hematin to repress porphyrin production. Combinations of these and other adjunctive agents and modalities may moderate the urgency presented by a failing organ, allowing orderly preparation for an optimal transplantation.
Photoprotectants
Beta-carotene is a scavenger of singlet-exited oxygen and is believed to interfere with the efficiency of porphyrin-sensitized photoxidative damage in the skin. Ingestion of beta-carotene at recommended doses produces carotenodermia after several weeks. Increasing tolerance of sunlight develops during this loading period. Tolerance diminishes over several weeks when treatment is stopped.
Vitamin A (Lumitene)
Exact mechanism of action not completely elucidated. Patient must become carotenemic before effects are observed. More than one internal light screen may be responsible for effects. May provide a limited level of photoprotection. Causes yellowing of skin (carotenoderma). Any photoprotection afforded increases slowly over 4- to 6-wk period after drug is commenced. When discontinued, skin color and benefit fade over several weeks.
Adult
120-300 mg/d PO in divided doses
Pediatric
30-120 mg PO in divided doses
Coadministration with vitamin A may result in additive toxic effects
Documented hypersensitivity; use by tobacco smokers may further increase risk of lung cancer
Pregnancy
B - Fetal risk not confirmed in studies in humans but has been shown in some studies in animals
Precautions
Caution in patients with renal or hepatic impairment; may increase risk for lung cancer in heavy smokers; may cause orange stools and diarrhea or loose stools at onset of therapy that tend to resolve with continued use
Antihistamines
H1-receptor antagonists modulate effects of histamine in skin. If taken prior to anticipated strong sunlight exposure that cannot be avoided, acute reactions may be attenuated to some extent; minimal benefit is expected if taken afterward.
Fexofenadine (Allegra)
Nonsedating second-generation medication with fewer adverse effects than first-generation medications. Competes with histamine for H1 receptors on GI tract, blood vessels, and respiratory tract, reducing hypersensitivity reactions. Does not sedate. Available in qd and bid preparations.
Adult
180 mg PO 2-3 h prior to sunlight exposure
Pediatric
<6 years: Not recommended
6-11 years: 30-60 mg PO 2-3 h prior to sunlight exposure
>12 years: Administer as in adults
Toxicity increases with coadministration of erythromycin and ketoconazole
Documented hypersensitivity
Pregnancy
C - Fetal risk revealed in studies in animals but not established or not studied in humans; may use if benefits outweigh risk to fetus
Precautions
No data available on use while breastfeeding; reduce dose in renal insufficiency
Enteric adsorbents
Agents that bind protoporphyrin in the intestinal lumen promote its excretion by interrupting enterohepatic recirculation, thereby reducing the porphyrin load presented to the liver for clearance.
Cholestyramine (Questran)
Polymeric resin that binds bile acids, porphyrins, and other molecules to form nonabsorbable complexes that are excreted unchanged in feces. Adsorbs many drugs and nutrients; long-term use requires proper timing of oral drugs and may warrant supplementation of vitamins D, E, A, and K.
Adult
4 g PO tid ac; may increase to 24 g/d PO in divided doses
Pediatric
<6 years: Not established
>6 years: 2 g PO bid initially; may increase up to 8 g/d PO divided tid/qid
Inhibits absorption of numerous drugs, including warfarin, thyroid hormone, amiodarone, NSAIDs, methotrexate, digitalis glycosides, glipizide, phenytoin, imipramine, niacin, methyldopa, tetracyclines, clofibrate, hydrocortisone, and penicillin G
Documented hypersensitivity
Pregnancy
B - Fetal risk not confirmed in studies in humans but has been shown in some studies in animals
Precautions
Caution in constipation and phenylketonuria
Activated charcoal (Actidose)
Prevents absorption by adsorbing porphyrin in intestine. Multidose charcoal may interrupt enterohepatic recirculation and enhance elimination by enterocapillary exsorption. Does not dissolve in water. Adsorbs many medications and nutrients; long-term use requires proper timing of oral drugs and may warrant supplementation of vitamins D, E, A, and K.
Adult
25-100 g or 1 g/kg PO susp in 4-8 oz of water
Pediatric
<1 year: Not recommended
>1 year: Administer as in adults
Effectiveness of other medications decreases with coadministration; do not mix charcoal with sherbet, milk, or ice cream (decreases absorptive properties)
Documented hypersensitivity
Pregnancy
C - Fetal risk revealed in studies in animals but not established or not studied in humans; may use if benefits outweigh risk to fetus
Precautions
Administer supplemental vitamins D, E, A, and K with long-term use
Antihistamines, H2 blocker
Produce blockade of H2 receptors.
Cimetidine (Tagamet)
H2 antagonist, which, when combined with an H1-type, may be useful in treating itching and flushing in urticaria. Porphyria-specific usage for inhibiting overproduction of porphyrins is experimental.
Adult
Experimental doses reported for inhibiting heme synthesis: 400 mg PO bid to 800 mg PO qid; not to exceed 2400 mg/d (recommended)
Pediatric
Not established for experimental use in porphyrias
Can increase blood levels of theophylline, warfarin, TCAs, triamterene, phenytoin, quinidine, propranolol, metronidazole, procainamide, and lidocaine
Documented hypersensitivity
Pregnancy
B - Fetal risk not confirmed in studies in humans but has been shown in some studies in animals
Precautions
Elderly patients may experience confusional states; may cause impotence and gynecomastia in young males; may increase levels of many drugs; adjust dose or discontinue treatment if changes in renal function occur
Gallstone dissolution agents
Increasing bile flow enhances secretion of protoporphyrin by the liver into the enteric tract and clearance from the body.
Ursodiol (Actigall)
Shown to promote bile flow in cholestatic conditions associated with a patent extrahepatic biliary system. Decreases cholesterol content of bile, therefore reduces bile stone and sludge formation.
Adult
10-15 mg/kg/d PO divided bid
Pediatric
20-30 mg/kg/d PO divided bid
Decreased effect with aluminum-containing antacids, cholestyramine, colestipol, clofibrate, and oral contraceptives
Documented hypersensitivity
Pregnancy
B - Fetal risk not confirmed in studies in humans but has been shown in some studies in animals
Precautions
Caution in patients with a nonvisualizing gallbladder