| Eruptive xanthoma = الاورام الصفر الاندفاعية |
|
|
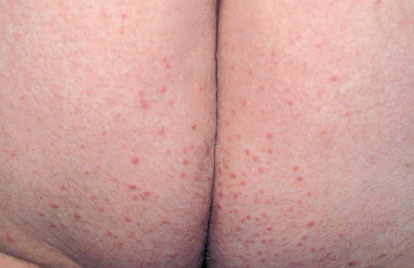
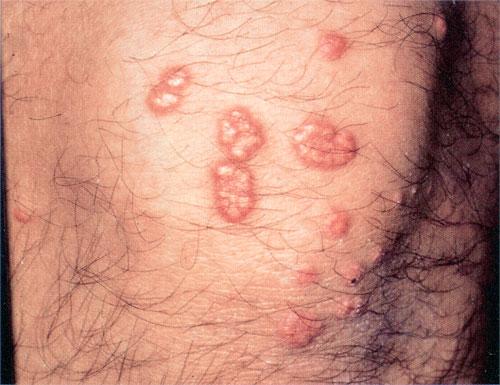
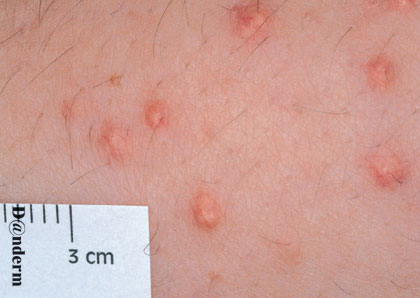
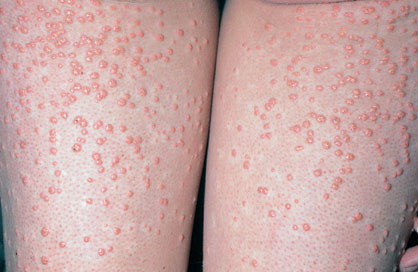
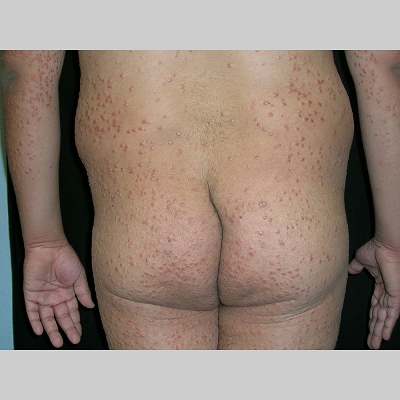
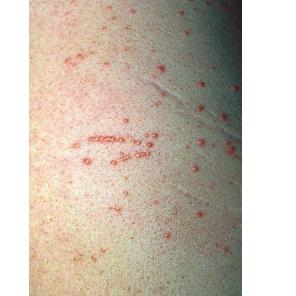
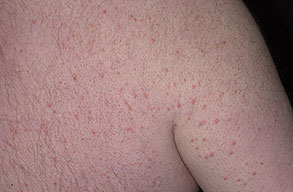
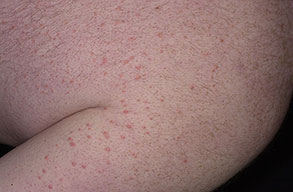
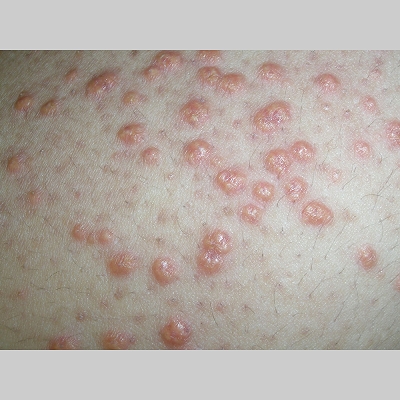
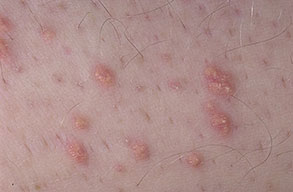
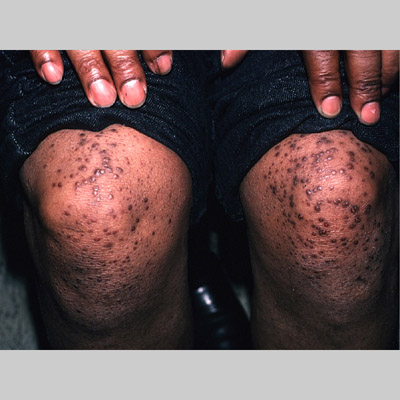
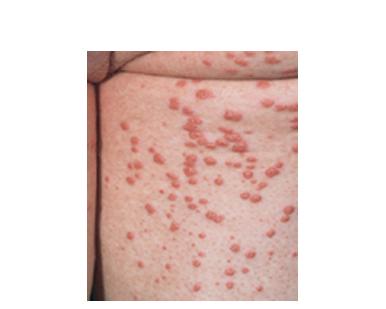
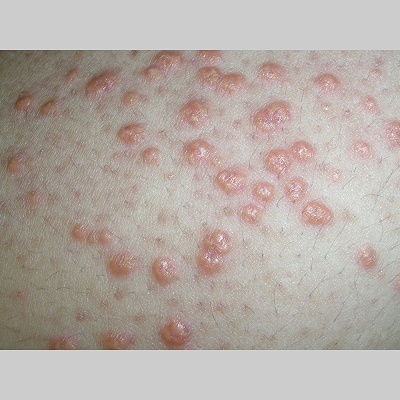
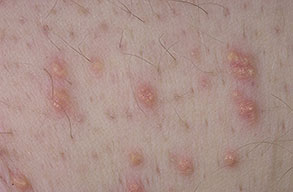
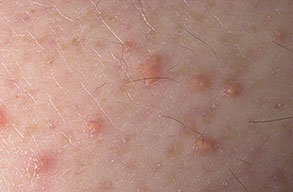
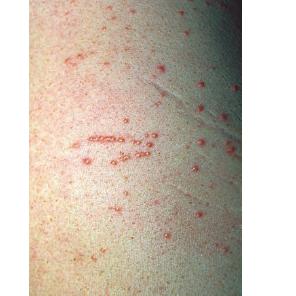
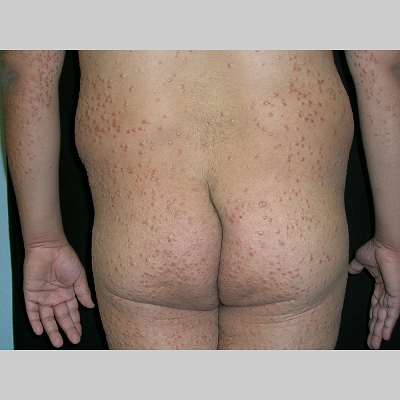
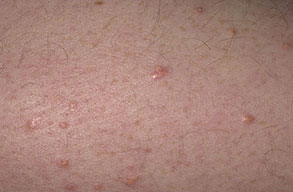
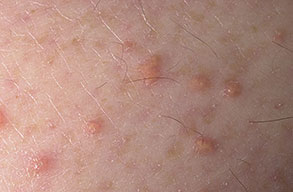
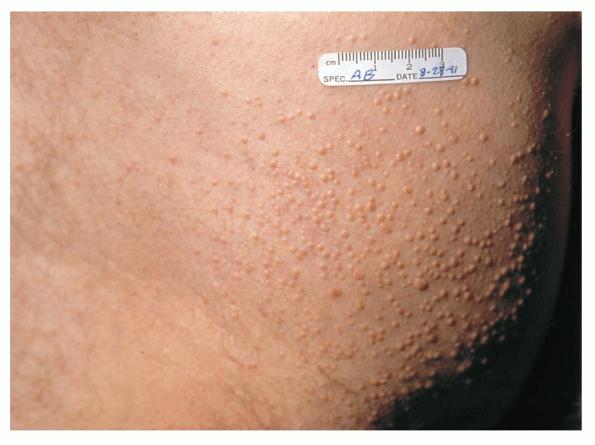
Eruptive xanthoma
Xanthomas are lesions characterized by accumulations of lipid-laden macrophages. Xanthomas can develop in the setting of altered systemic lipid metabolism or as a result of local cell dysfunction. PathophysiologyLipids are insoluble in water; therefore, they are transported as complexes of lipoproteins and specific apoproteins. These proteins also serve as ligands to specific receptors, they facilitate transmembrane transport, and they regulate enzymatic activity. Lipoproteins may be classified according to their density, as follows: chylomicrons, very-low-density lipoproteins (VLDL), intermediate-density lipoproteins (IDL), low-density lipoproteins (LDL), and high-density lipoproteins (HDL). Lipoproteins may also be separated by electrophoresis into beta (LDL), prebeta (VLDL), and alpha (HDL) lipoproteins. Beta-VLDL (IDL) can be determined by ultracentrifugation and electrophoresis. The metabolic pathways of lipoproteins can be divided into exogenous and endogenous pathways. The exogenous lipoprotein pathway refers to the metabolism of intestinal lipoproteins, the triglyceride-rich chylomicrons, primarily formed in response to dietary fat. The endogenous lipoprotein pathway refers to lipoproteins and apoproteins that are synthesized in tissues other than the intestines, predominantly in the liver. The liver secretes the triglyceride-rich VLDL that contains apoproteins B-100, C-II, and E into the circulation. In the peripheral tissues, particularly adipose and muscle tissue, VLDL is cleaved by lipoprotein lipase (LPL), extracting most of the triglycerides and forming an IDL that contains apoproteins B-100 and E. IDL can be taken up by the liver through the LDL receptor, or it can be converted to the cholesterol-rich LDL that contains apoprotein B-100. LDL is removed from the circulation primarily by the liver through the LDL receptor. HDL particles that contain apoproteins A-I and A-II interact with other lipoproteins, particularly VLDL and LDL, through lipolysis and the action of lecithin cholesterol acyltransferase (LCAT) enzyme. The main role of HDL is to accept cholesterol and to transport it back to the liver (reverse cholesterol transport). Lipoprotein (a) (Lp[a]) consists of an LDL-like particle with apoprotein B and a side chain of a highly glycosylated protein. Lp(a) has a role not only in atherogenesis but also in thrombogenesis because of its homology with plasminogen. Alterations in lipoproteins result either from genetic mutations that yield defective apolipoproteins (primary hyperlipoproteinemia) or from some other underlying systemic disorder, such as diabetes mellitus, hypothyroidism, or nephrotic syndrome (secondary hyperlipoproteinemia). The biochemical and genetic basis for the inherited disorders of lipid and lipoprotein metabolism differ considerably. Traditionally, hyperlipidemias have been classified according to 6 phenotypes described by Fredrickson. These phenotypes are based on the electrophoretic patterns of lipoprotein level elevations that occur in patients with hyperlipoproteinemia. In recent years, the understanding of the genetic and biochemical basis of these disorders has revealed a large and diverse group of diseases, many of which have similar clinical expressions, exposing the limitations of the Fredrickson classification system. Despite the system's shortcomings, Fredrickson phenotypes are a useful tool for the discussion of these disorders. The understanding of the pathophysiology of these defects provides a basis for diagnosis and treatment. Familial lipoprotein lipase deficiency is an example of a primary disorder in which a deficiency of lipoprotein lipase in tissue leads to a type I pattern of hyperlipidemia, with a massive accumulation of chylomicrons in the plasma. This effect results in a severe elevation of plasma triglyceride levels. Plasma cholesterol levels are not usually elevated. Patients with type I may present in early childhood, often with acute pancreatitis. Eruptive xanthomas are the most characteristic skin manifestation of this disorder. Cholesterol is bound to apolipoprotein B-100 as LDL in interstitial fluid. Cells may acquire cholesterol via an LDL receptor on the cell membrane. Familial LDL receptor deficiency and familial defective apoprotein B-100 are examples of primary defects that can lead to the accumulation of LDL, which corresponds to a type IIa pattern of hyperlipidemia. Plasma cholesterol levels are severely elevated, but plasma triglyceride levels are typically normal. Patients with type IIa have severe atherosclerosis and may present with tendinous or tuberous xanthomas as well as xanthelasmas. The type IIb pattern is characterized by the accumulation of both LDL and VLDL, with variable elevations of both triglyceride levels and cholesterol levels in the plasma. Patients with familial combined hyperlipoproteinemia have such a pattern of hyperlipidemia, but a specific genetic defect has not been established. Patients with type IIb may present as adults with tendinous or tuberous xanthomas as well as xanthelasmas. Type III hyperlipidemia is characterized by the accumulation of IDL (beta-VLDL), which is manifested by increases in both triglyceride levels and cholesterol levels in the plasma. A genetic basis for the primary disorder, familial dysbetalipoproteinemia, has been well established. Various mutations of apoprotein E impair its ability to bind to the IDL receptor. Patients with type III present as adults with premature atherosclerosis and xanthomas, particularly plane (palmar) xanthomas.1 Familial hypertriglyceridemia is an example of a primary defect resulting in type IV hyperlipidemia. Accumulation of VLDL causes severe elevations of plasma triglyceride levels. Plasma cholesterol levels are typically normal. A definitive molecular defect has not been established. Patients with type IV may present with eruptive xanthomas. Genetic defects of the apolipoprotein C-II gene result in the accumulation of chylomicrons and VLDL, which is the type V pattern of hyperlipidemia. Patients with this type have severe elevations of triglyceride levels in the plasma. These patients, like those with lipoprotein lipase deficiency, may present in early childhood with acute pancreatitis and eruptive xanthomas. Decreased synthesis of HDL due to decreased formation of apoprotein A-I and apoprotein C-III leads to decreased reversed cholesterol transport, resulting in increased LDL levels, premature coronary artery disease, and plane xanthomas. Hyperlipidemia is also related to a variety of secondary causes. Secondary hypercholesterolemia can be found in pregnancy, hypothyroidism, cholestasis, and acute intermittent porphyria. Secondary hypertriglyceridemia can be associated with oral contraceptive use, diabetes mellitus, alcoholism, pancreatitis, gout, sepsis due to gram-negative bacterial organisms, and type I glycogen storage disease. Combined hypercholesterolemia and hypertriglyceridemia can be found in nephrotic syndrome, chronic renal failure, and steroid immunosuppressive therapy.
History
PhysicalCutaneous xanthomas associated with hyperlipidemia can be clinically subdivided into xanthelasma palpebrarum, tuberous xanthoma, tendinous xanthoma, eruptive xanthoma, plane xanthoma, and generalized plane xanthoma. Xanthoma disseminatum and verruciform xanthoma are usually not associated with hyperlipidemia.
Laboratory Studies
Histologic FindingsChanges in the skin and the tendons are characterized by the presence of vacuolated macrophages (foamy macrophages). These macrophages are filled with lipid droplets, which are dissolved and removed from the tissue during histologic processing. Lipid stains are of no use in routinely processed tissue. In contrast, frozen sections can be stained with lipid stains. Foamy histiocytes usually have 1 nucleus, but multinucleated histiocytes (Touton giant cells) are often identified. Eruptive xanthomas may contain infiltrates of lymphocytes and typically contain extracellular lipid. Xanthelasma differs from other xanthomas because of the superficial location of the foamy cells and the characteristic appearance of eyelid skin. Tendinous and tuberous xanthomas can contain prominent fibrosis and occasional cholesterol clefts. Verruciform xanthoma demonstrates prominent hyperplasia of the squamous epithelium and foamy macrophages within dermal papillae.
Medical CareXanthomas not always associated with underlying hyperlipidemia, but when they are, diagnosing and treating underlying lipid disorders is necessary to decrease the size of the xanthomas and to prevent the risks of atherosclerosis. Treatment of the hyperlipidemia initially consists of diet and lipid-lowering agents such as statins, fibrates, bile acid–binding resins, probucol, or nicotinic acid. The lipid-lowering effects of these agents have been well documented, but few studies mention the efficacy of these drugs for resolving xanthomas. Eruptive xanthomas usually resolve within weeks of initiating systemic treatment and tuberous xanthomas usually resolve after months, but tendinous xanthomas take years to resolve or may persist indefinitely.
Surgical CareSurgery or locally destructive modalities can be used for idiopathic or unresponsive xanthomas. Xanthelasmas are often treated with topical trichloroacetic acid, electrodesiccation, laser therapy, and excision; however, recurrences can occur. Care must be taken to protect the eyes during any procedure used to treat xanthelasma. Such procedures should be performed only by individuals who are thoroughly familiar with and skilled in the procedure.
|