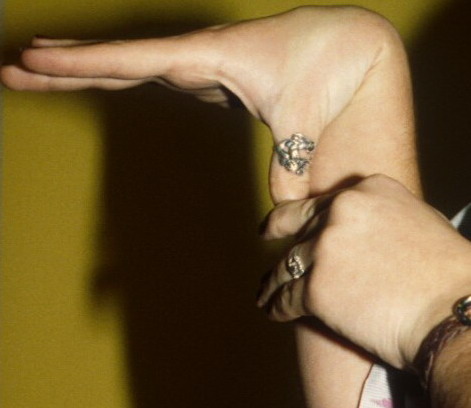▪ EHLERS-DANLOS SYNDROMES
The Ehlers-Danlos syndromes (EDSs) are a heterogeneous group of disorders characterized by hypermobile joints, hyperextensible skin, and fragile tissues that are extremely susceptible to trauma. As many as 1 in 5000 individuals may be affected by some form of the disease.The seven sub-types of EDS have been categorized based on clinical and corresponding molecular pathogenetic findings in collagen types I, III, and V, or processing enzymes. These collagen types are all fibrillar types of collagen with cross-banding that are expressed in different locations in the skin and in other tissues . Pathogenic alterations of fibrillar collagens in EDS result from deficient collagen-processing enzymes, haploinsufficiency, or mutations in collagen α chains. A uniform finding in all types of EDS is generalized joint hypermobility, which can be assessed clinically according to the Beighton scale
Classical Type
EPIDEMIOLOGY
The classical form of EDS is one of the most common, occurring in 1 in 10,000 to 20,000 infants.9
ETIOLOGY AND PATHOGENESIS
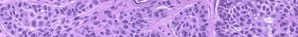
The classical type of EDS is characterized by an alteration in type V fibrillary collagen and is inherited in an autosomal dominant (AD) fashion. Mutations in COL5A1 and COL5A2 have been shown to cause this variant in up to 50 percent of cases. The abnormal allele leads to dominant-negative effects or haploinsufficiency, depending on the mutation. At least one other locus is involved, because several families are discordant for linkage to both COL5A1 and COL5A2. A few patients have shown mutations in COL1A1, a gene more commonly mutated in osteogenesis imperfecta (OI; see Osteogenesis Imperfecta).
Whereas COL5A1 is located on the long arm of chromosome 2q31, the COLA5A2 gene resides on the long arm of chromosome 9q34.2-34.3. Although collagen V is not abundant in skin, tendons, or ligaments, the clinical phenotype attests to its important structural role.
EHLERS-DANLOS SYNDROMES AT A GLANCE
- Combined incidence of almost 1 in 5000 persons.
- Seven sub-types.
- Most commonly autosomal dominant (classical and hypermobile types).
- The gene encoding collagen V is most often affected.
- Cutaneous features include soft, velvety skin that bruises easily and wounds that heal as thin, atrophic, gaping scars.
- Extracutaneous manifestations include hypermobile joints with frequent dislocations, problems with pregnancy and delivery, and, less commonly, cardiovascular manifestations, particularly aortic root dilatation.
- Information for patients and professionals at http://www.ehlers-danlos.org and http://www.ednf.org.
The Ehlers-Danlos Syndromes: Clinical Sub-Types and Associated Defects
|
|
VILLEFRANCHE TYPE/(OMIM)
|
CLINICAL FEATURES
|
INHERITANCE
|
PROTEIN/(GENE DEFECT)
|
|
Classical/(130000 and 130010)
|
Hyperextensible skin; easy bruising; wide, atrophic scars; hypermobile joints
|
AD
|
Collagen type V/(COL5A1, COL5A2)
|
|
Hypermobility/(130020)
|
Smooth, velvety skin; joint hypermobility
|
AD/AR
|
Unclear for most; collagen type III; tenascin XB/(COL3A1; TNXB)a
|
|
Vascular/(130050)
|
Thin, translucent skin with easy bruising; arterial and visceral rupture; typical facies
|
AD
|
Collagen type III/(COL3A1)
|
|
Kyphoscoliosis/(225400 and 229200)
|
Atrophic scars, easy bruising; neonatal hypotonia; scoliosis; ocular rupture; marfanoid habitus
|
AR
|
Lysyl hydroxylase/(PLOD1)
|
|
Arthrochalasia/(130060)
|
Hyperextensible and fragile skin; severe joint hypermobility; congenital hip dislocation
|
AD
|
Collagen type I/(COL1A1; COL1A2)
|
|
Dermatosparaxis/(225410)
|
Severely fragile, sagging, redundant skin; hernias and premature rupture of fetal membranes
|
AR
|
Procollagen I N-peptidase/(ADAMTS2)
|
|
Other typesa
|
|
|
|
|
|
Progeroid variant190/(130070)
|
Wrinkled, loose facial skin, curly fine hair, scanty eyebrows and eyelashes
|
AR
|
Due to mutations in galactosyltransferase
|
|
AD = autosomal dominant; AR = autosomal recessive; OMIM = Online Mendelian Inheritance in Man.
|
|
a Few reported cases.
|
| |
|
|
|
|
|
Ultrastructural findings show thickened collagen fibrils in skin,with an approximate 25 percent increase in diameter, underscoring the function of collagen V in limiting fibril diameter, as shown in vitro.22 This effect is thought to be due to the negative charge of the amino terminus of α1(V), conferred by abundant tyrosine residues, which inhibits fibril growth. Another hallmark of classical EDS is the presence of rare composite fibrils also known as “collagen cauliflowers.” These abnormalities are present in less than 5 percent of fibrils, suggesting that other factors may be responsible for the severe clinical phenotype, such as the arrangement of collagen fibrils in tissue. This disorganized arrangement of collagen fibrils and their interaction with other extracellular matrix (ECM) proteins is thought to cause the altered biomechanical properties of EDS skin.24
CLINICAL FINDINGS
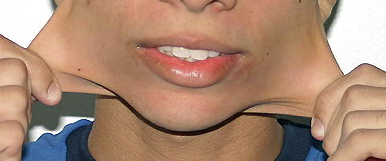
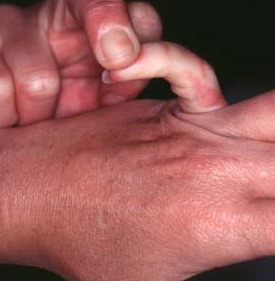
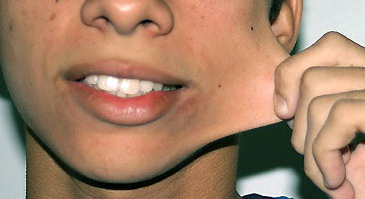
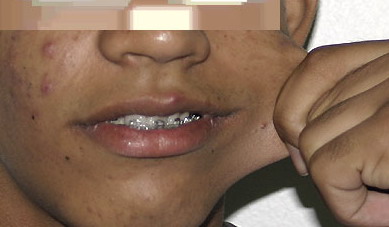
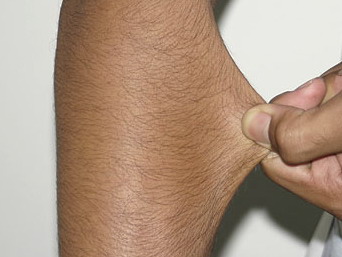
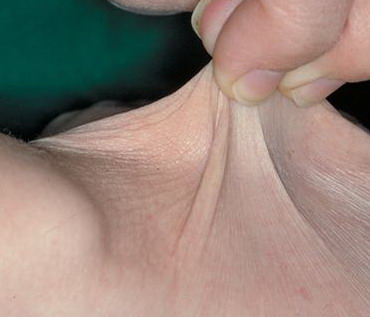
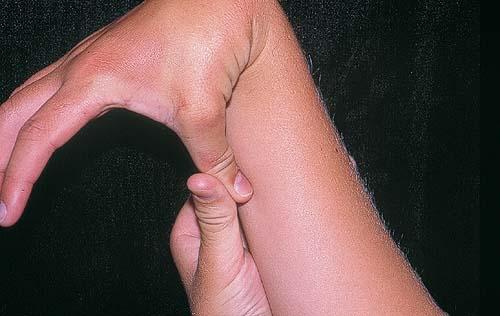
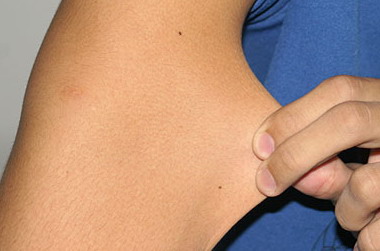
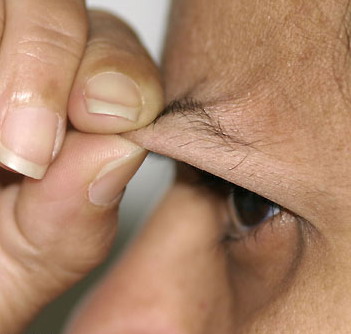
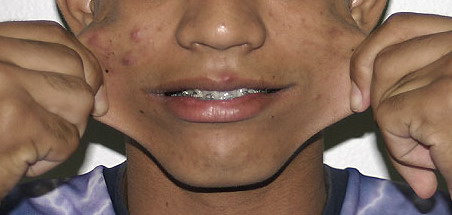
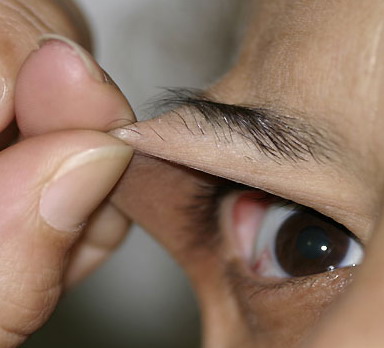
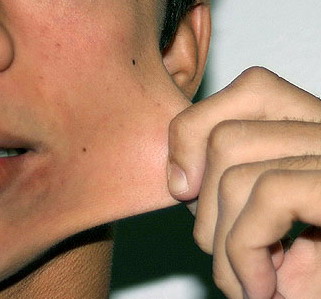
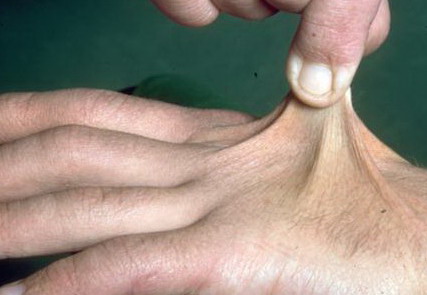
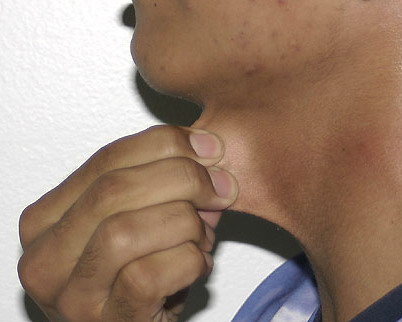
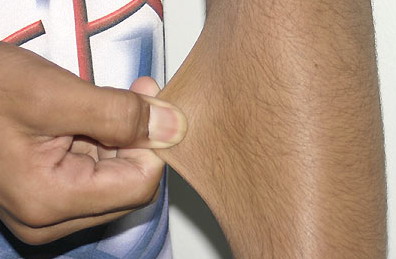
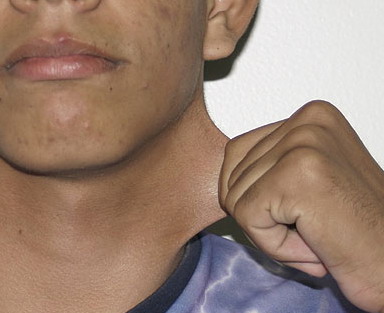
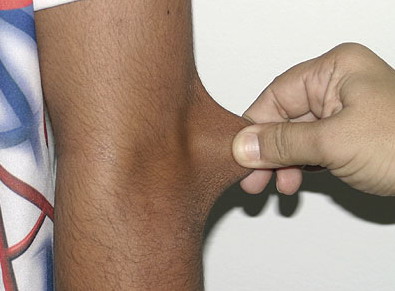
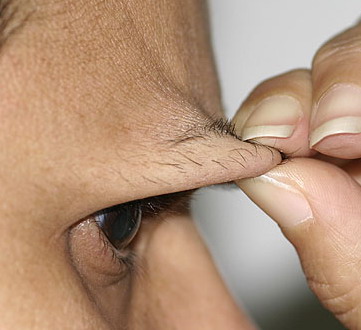
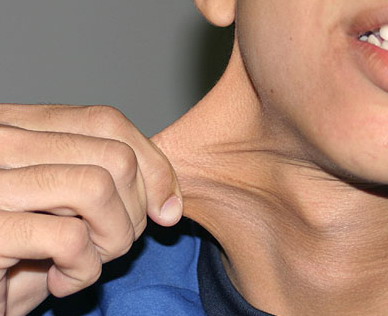
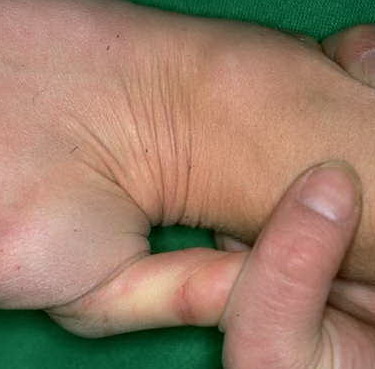
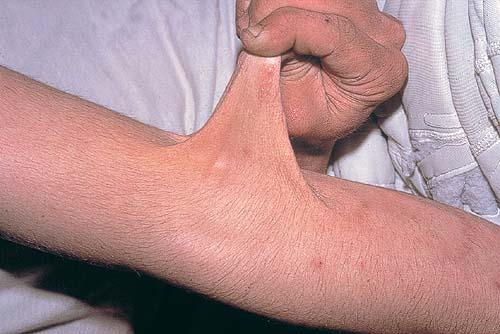
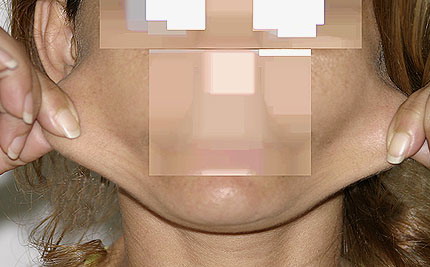
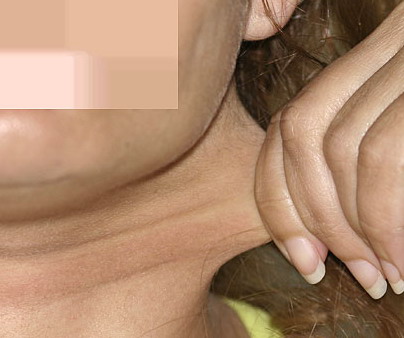
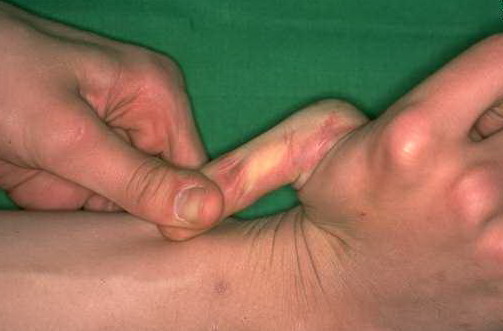
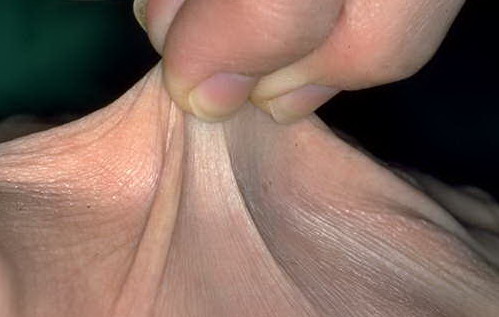
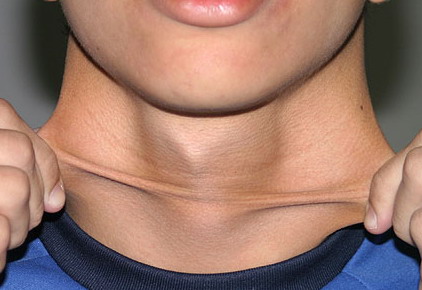
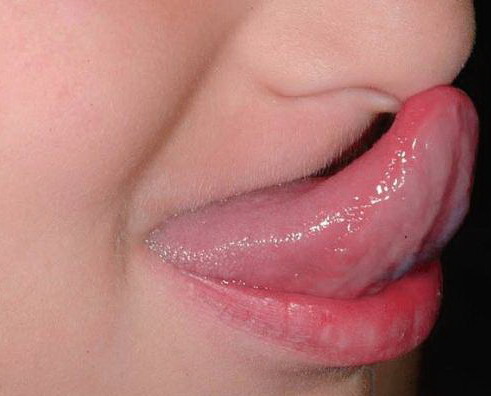
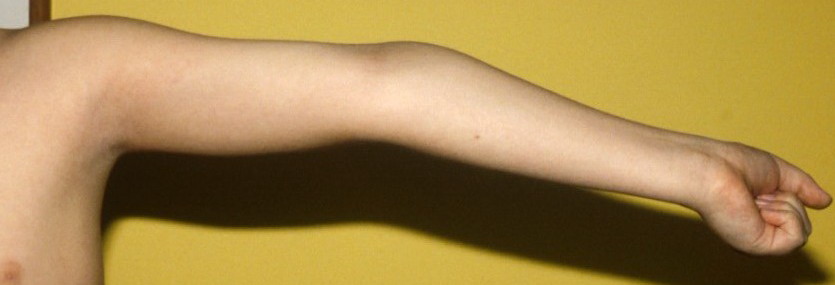
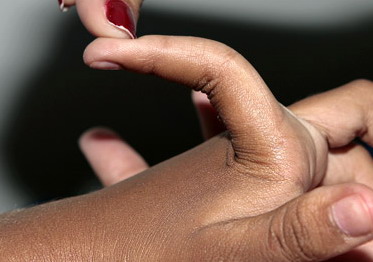
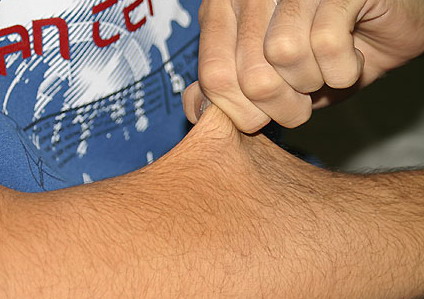
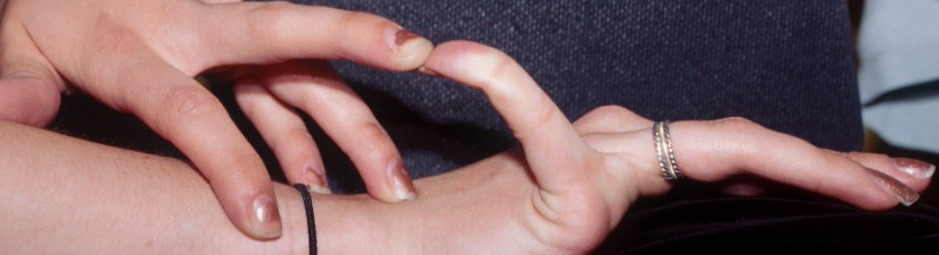
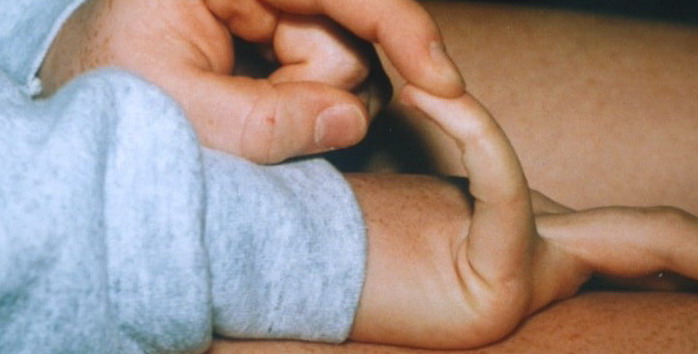
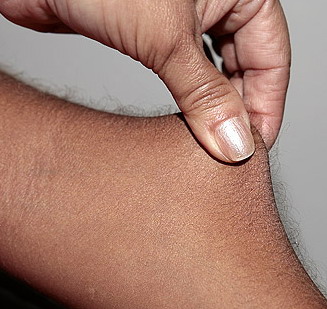
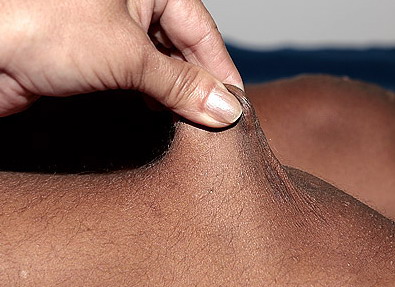
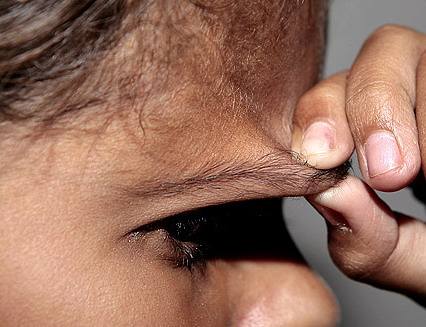
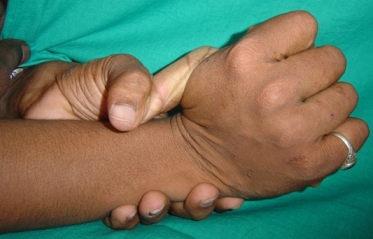
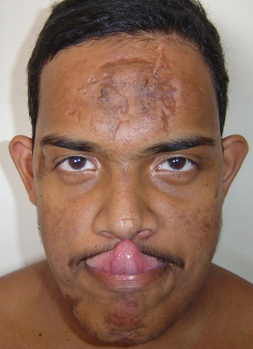
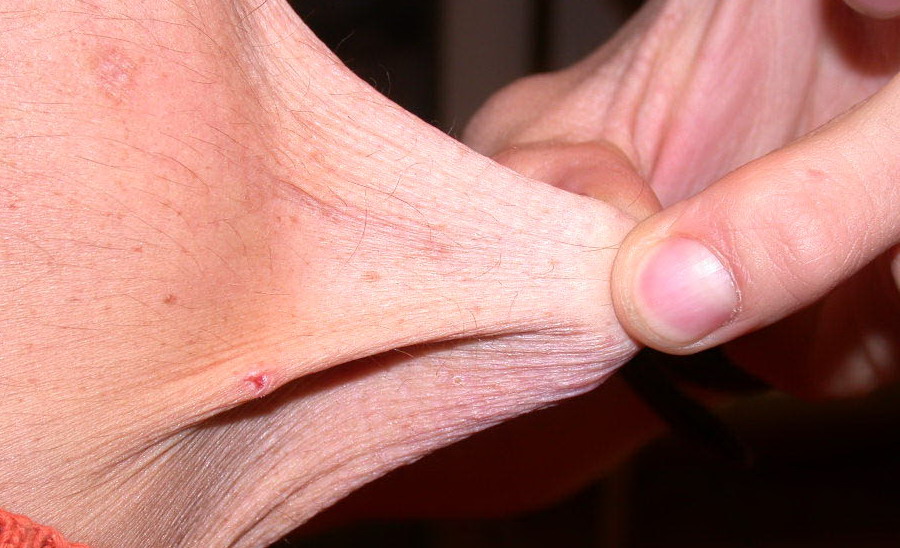
Classical EDS is characterized by joint laxity, hyperextensibility of skin, and poor wound healing. The skin manifestations can vary in severity from mild to severe; milder forms were previously termed the “mitis” type, or type II, EDS. The skin is soft, velvety, and can be stretched easily . It is not lax, except in late stages. Skin hyperextensibility should be determined at the volar surface of the forearm or some other site that is not subjected to mechanical forces or scarring. Approximately 50 percent of patients with Ehlers-Danlos of the classic and hypermobile types can touch the tip of their nose with their tongue (Gorlin's sign), in contrast to 10 percent of individuals who do not have EDS
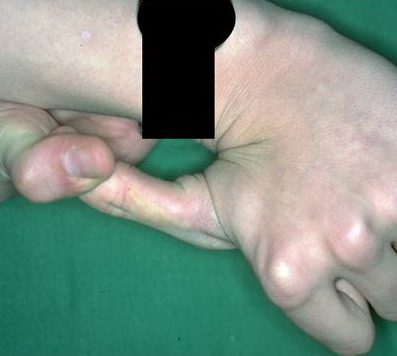
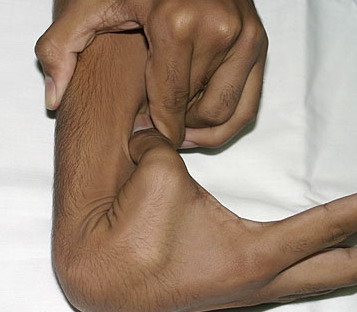
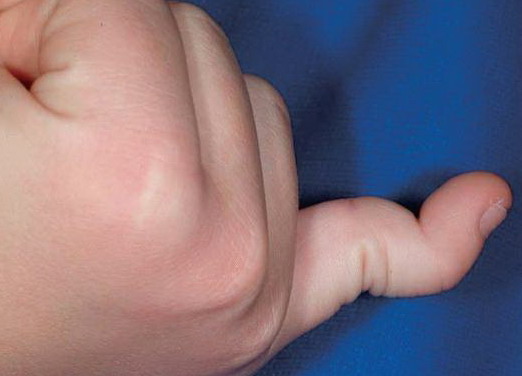
Beighton Criteria for Joint Hypermobility
|
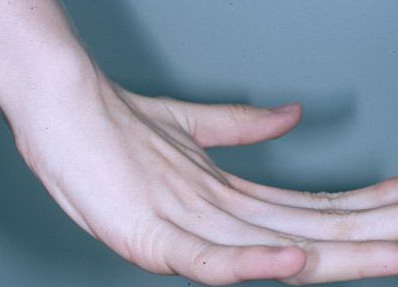
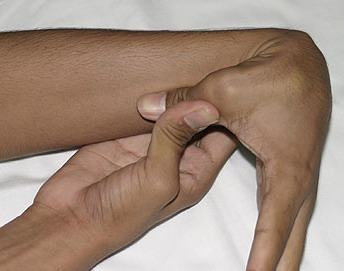
- Passive dorsiflexion of the fifth finger > 90 degrees
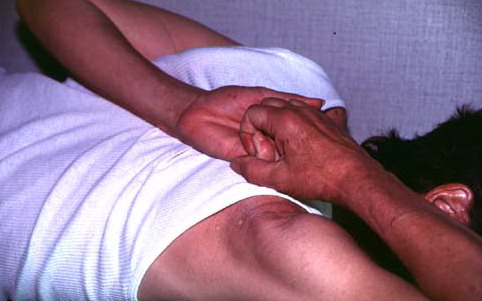
- Passive apposition of the thumbs to the flexor aspect of the forearm (Beighton sign)
- Hyperextension of the elbow > 10 degrees
- Hyperextension of the knees > 10 degrees
- Ability of the palms to completely touch the floor during forward flexion of the trunk with knees fully extended
|
|
Numbers 1-4 are scored for each side, so that a maximal score of two is possible if both left and right sides show criteria; number 5 is scored as 1. A score of ≥ 5/9 is hypermobile.
|
|
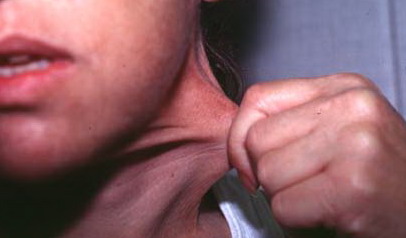
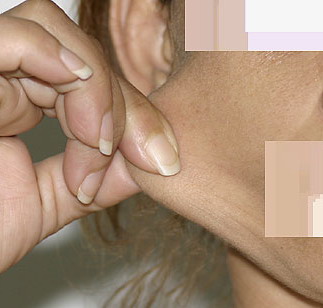
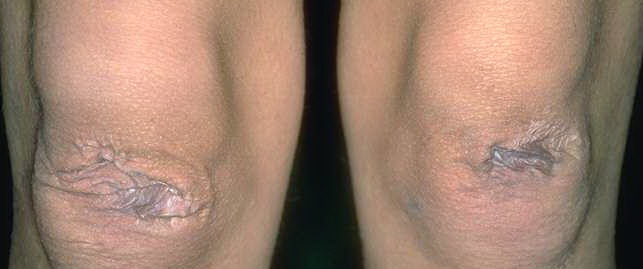
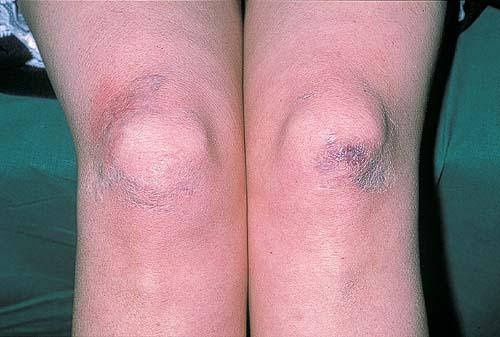
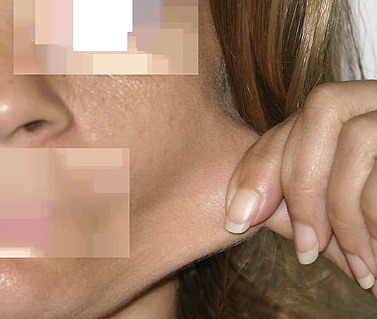
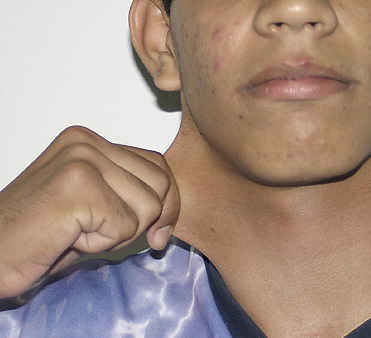
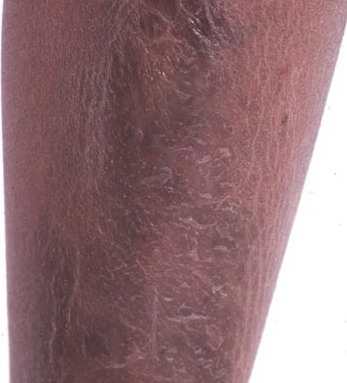
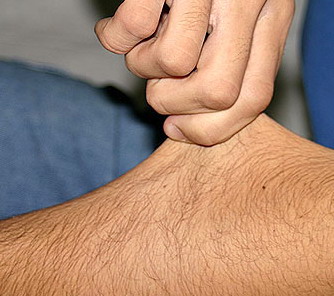
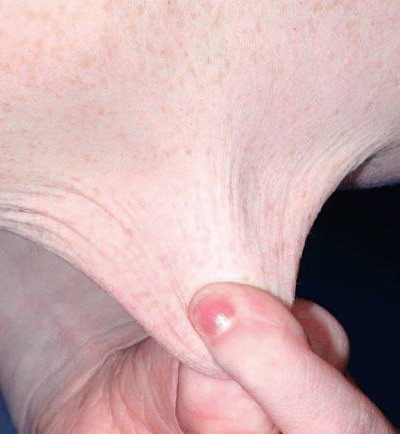
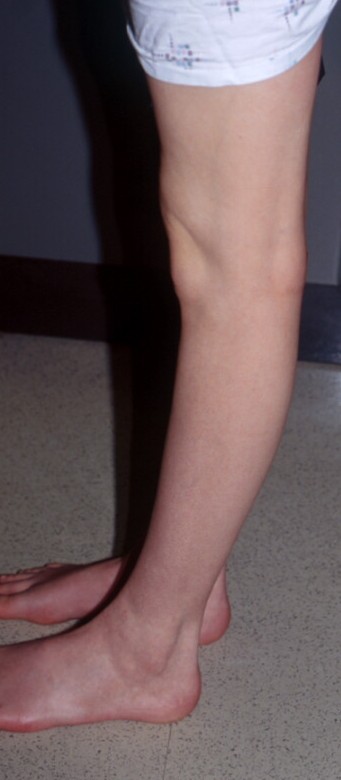
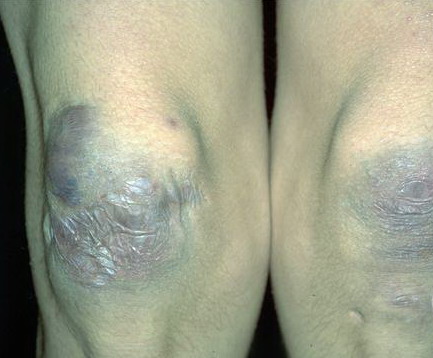
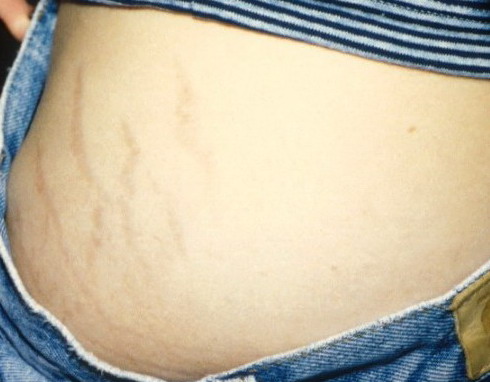
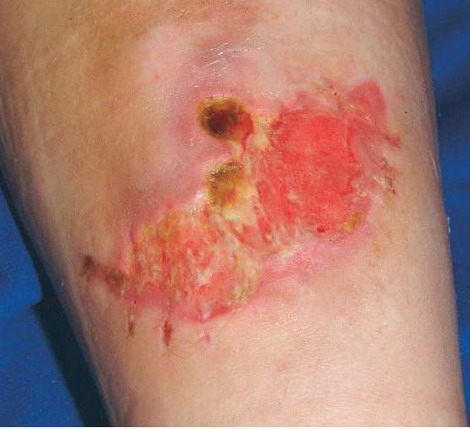
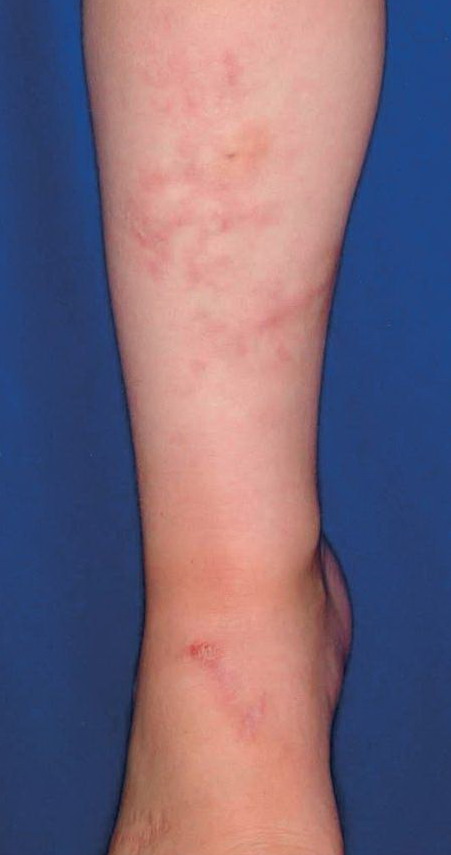
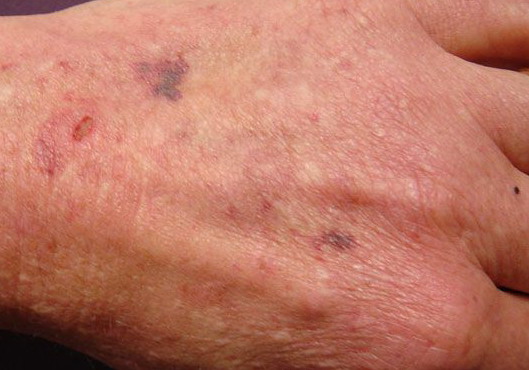
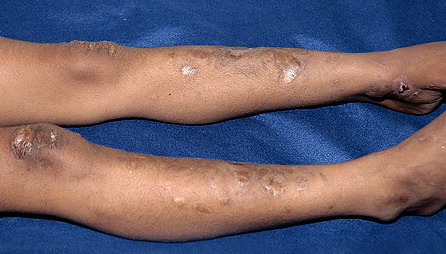
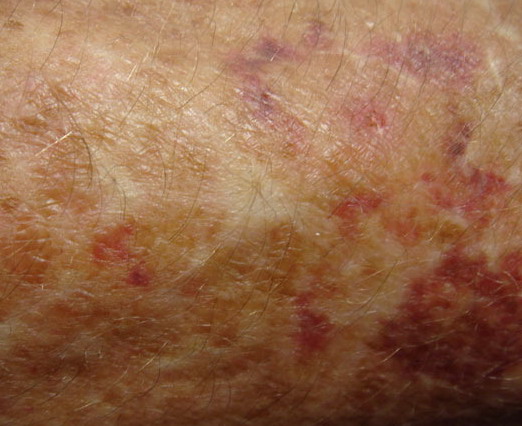
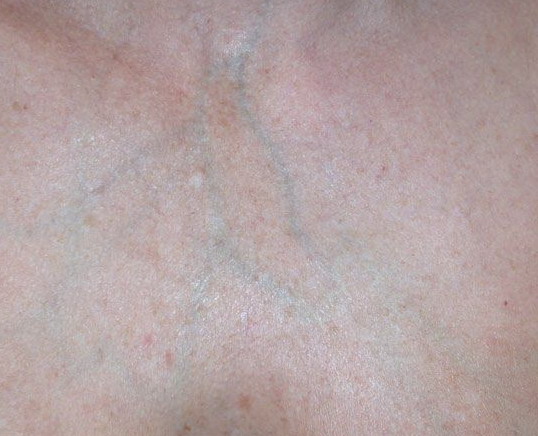
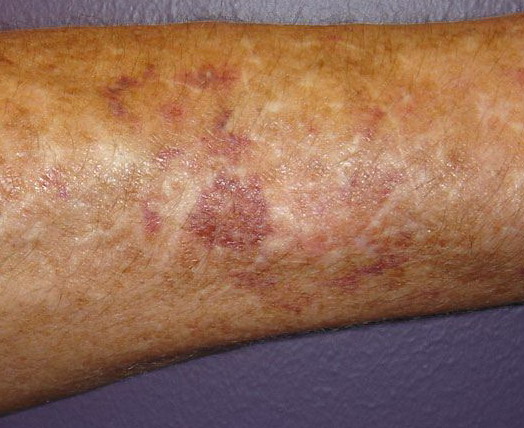
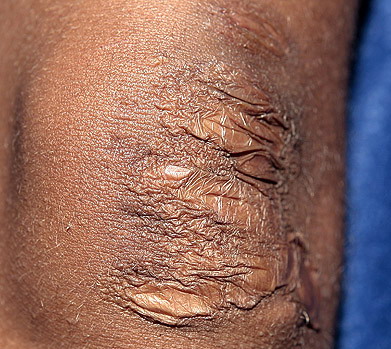
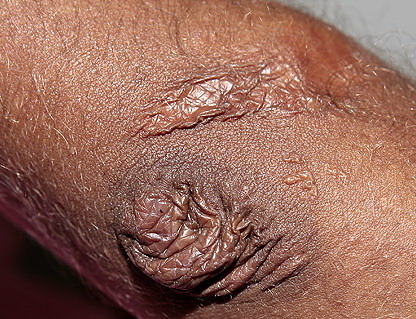
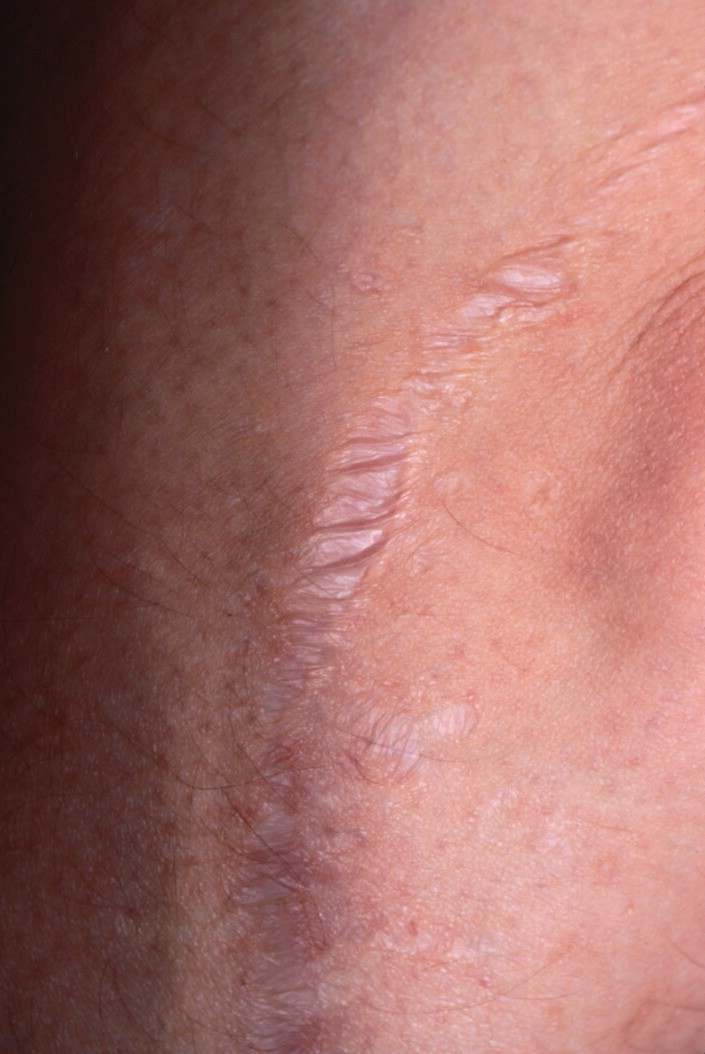
The dermis is fragile and easily bruised. The shins show persistent discoloration, often beginning in early childhood . When the skin splits from trauma, it is relatively painless and does not bleed excessively, but the wounds tend to gape. The wound margins tend to retract, heal slowly, and often become infected. Dehiscence is common, and complete wound breakdown
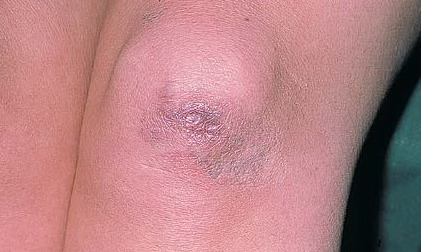
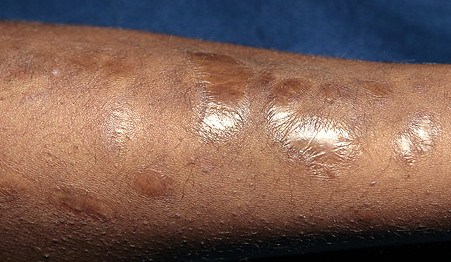
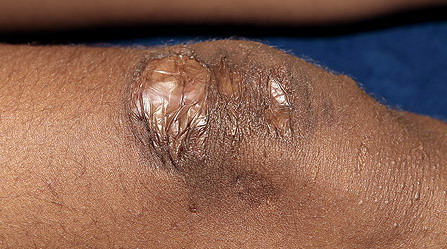
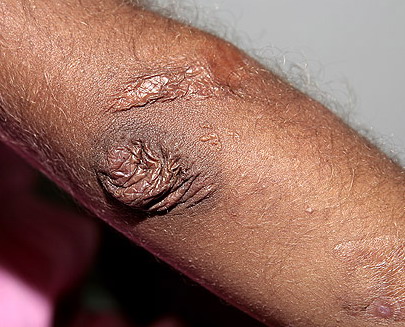
may require repeated suturing or healing by secondary intention Alternatively, the skin may appear to hold the stitches well initially, but the wound may fall apart after sutures are removed. Scars after trauma or surgical procedures are thin, papyraceous (having the consistency of paper), and may stretch considerably after healing . The more severely affected individuals have scars with a characteristic “fish mouth” or “cigarette paper” appearance. Thin, atrophic, darkly pigmented scars form as a consequence of intradermal or subdermal hematomatas, and occur mainly at pressure points. Molluscoid pseudotumors, fleshy lesions associated with scars, are present at the extensor surfaces of joints, in the foot, and on the shins. Spheroids are well-defined, mobile, subcutaneous indurated nodules that occur at sites of recurrent trauma and can be confused with subcutaneous granuloma annulare. Affected persons may have pressure-induced herniation of subcutaneous fat on the wrists or on the medial or lateral aspect of the heels, evident when the patient is standing (piezogenic pedal papules). Hiatal hernia, post-operative hernias, and anal prolapse have been noted as manifestations of the tissue
hyperextensibility and fragility.
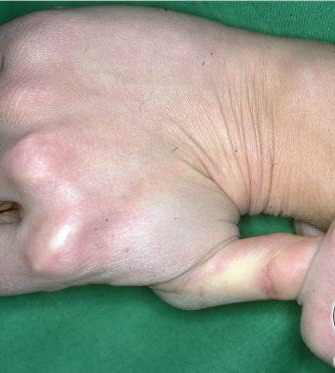
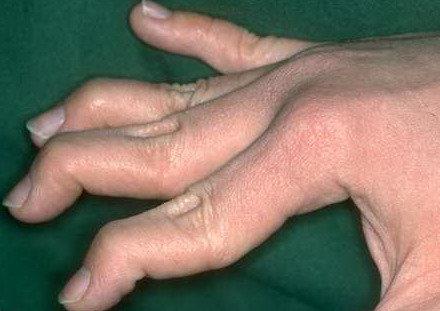
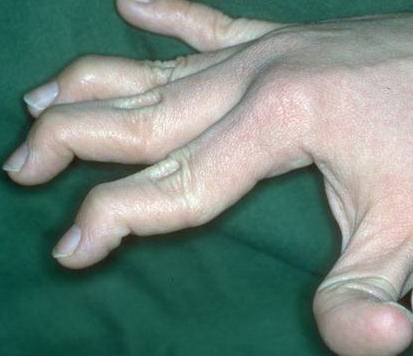
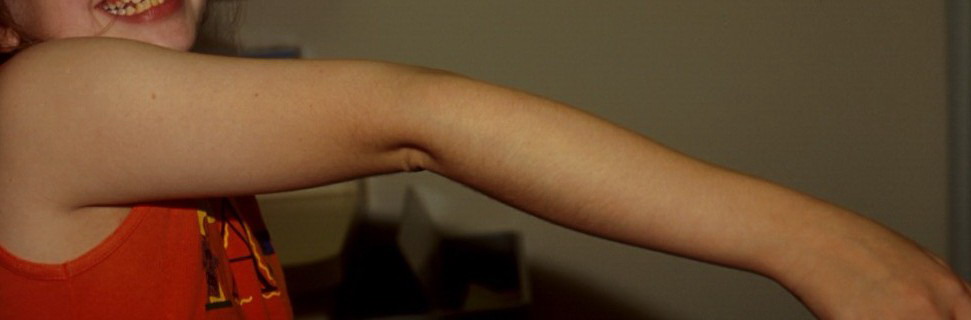
Musculoskeletal features seen in classical EDS include joint hyperextensibility in all patients and a fairly high frequency of scoliosis and pes planus.26 Patients tend to have “double-jointed” fingers and frequent sprains or subluxation of larger joints , either spontaneously or after slight trauma. Affected individuals complain of chronic joint and limb pain despite normal skeletal radiographs.27 The joint hypermobility can lead to the onset of osteoarthritis in the third or fourth decade. Muscle hypotonia and delayed gross motor development have been described.
Dyspareunia and sexual dysfunction have been reported, and the signs of classical EDS may be aggravated by pregnancy. Forty percent of affected individuals and 21 percent of babies of affected mothers are born prematurely, typically between 32 and 37 weeks, owing to premature rupture of fetal membranes or infection of the chorioamnionic membrane. Fetuses with classical EDS may exhibit retarded growth, hernias, and joint dislocations. A significant number of individuals with EDS have abnormal cardiac echocardiographic parameters, showing aberrant vessels and incompetent valves.31 Aortic root dilatation has been described in 29 percent of patients with the classical and hypermobile forms of EDS, especially in the former, and may lead to rupture. The most commonly affected site is the sinus of Valsalva. Initial screening of all EDS patients by echocardiography is recommended. Aortic valve incompetence, aortic rupture, and dissection occur far less commonly than in Marfan syndrome. β Blocker therapy with agents such as atenolol has been attempted in individual cases, but too few data exist to determine whether therapy is indicated or effective. Mitral valve prolapse (MVP) occurs no more frequently than in the general population.
The diagnosis is confirmed by clinical examination, family history, and the identification of mutations in COL5A1 or COL5A2, a test not commercially available.