atlas of dermatology
|
atlas of histopathology
|
· A heterogeneous group of disorders with altered immune responses selective to candida · Recurrent, progressive candidal infections of the skin, nails, and mucous membranes · May be associated with the later development of endocrinopathy [(APECED) autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy]
ETIOLOGY AND PATHOGENESIS
The clinical features of CMC may be seen in a variety of immunologic disorders, all characterized by ineffective defense mechanisms against Candida. In general, the patients with greater severity and an earlier onset of cutaneous candidal infections have more severe immunologic alterations. CMC patients have shown general dysregulation of interleukin 12 (IL-12), IL-6, and interferon-γ (IFN-γ) production,resulting in an inability to mount a cell-mediated response to clear candidal organisms. Chronic infections result in production of high levels of inflammatory cytokines (IL-6) followed by anti-inflammatory cytokines (IL-10) that further reduce the production of T helper 1 (Th1)-inducing cytokines via a positive feedback loop. Humoral immunity appears normal in most patients and 25 percent to 35 percent of patients with CMC have no demonstrable immunologic defects. Many patients with CMC have associated APECED syndrome, owing to mutations in the autoimmune regulator (AIRE) gene which maps to 21q22.3 and encodes a DNA transcription factor.41 Mice that are deficient in AIRE do not delete organ-specific T cells in the thymus, thus promoting the development of autoimmune disease.42 The reason for susceptibility to mucocutaneous
CLINICAL FINDINGS
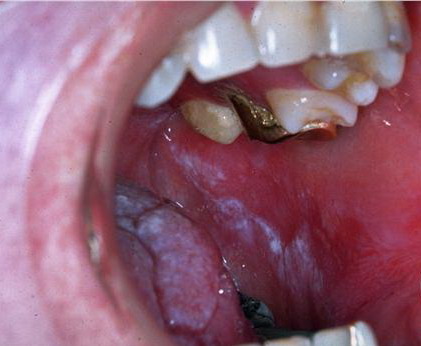
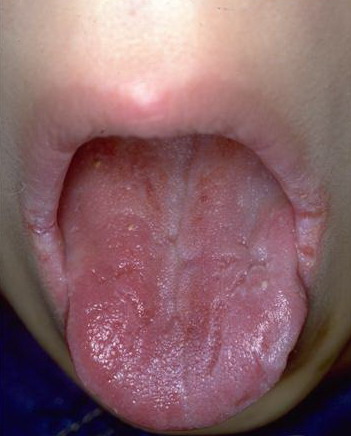
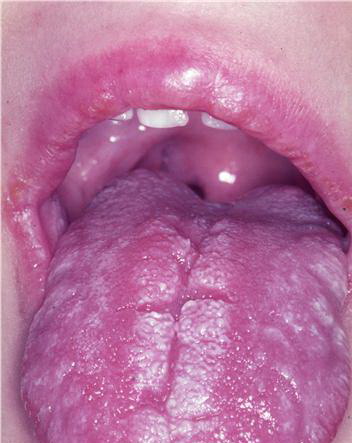
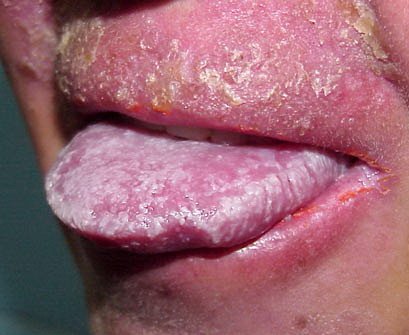
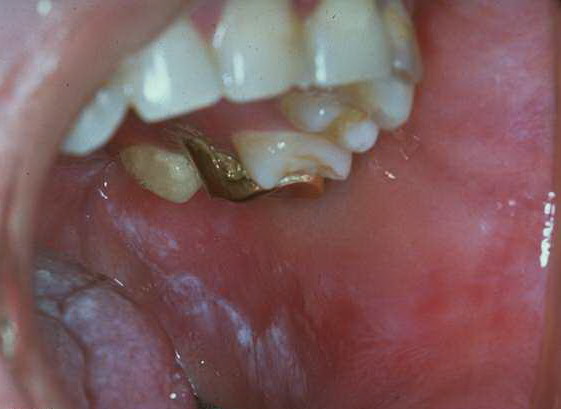
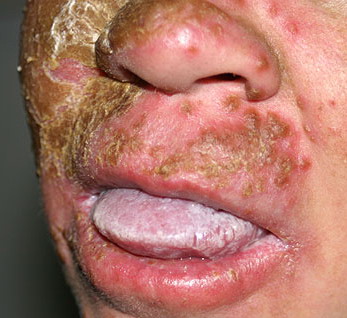
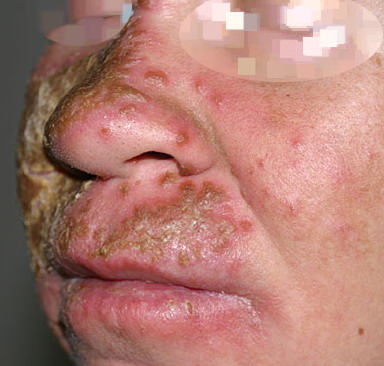
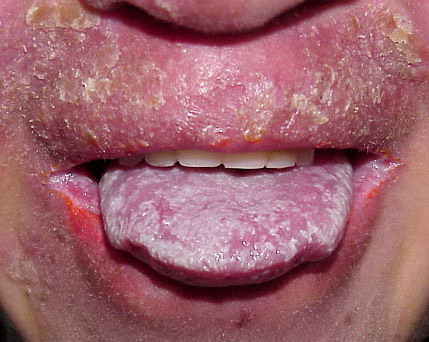
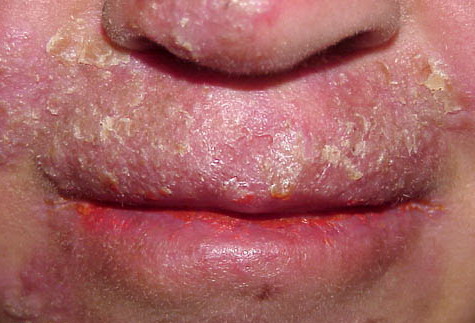
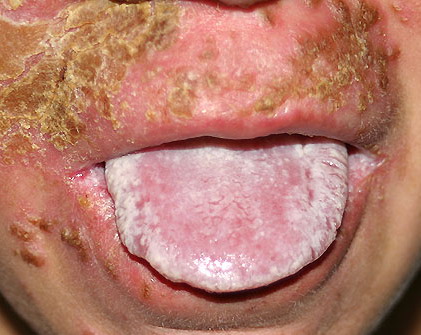
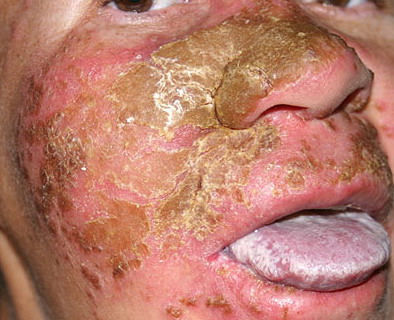
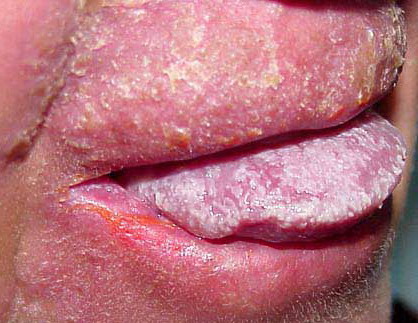
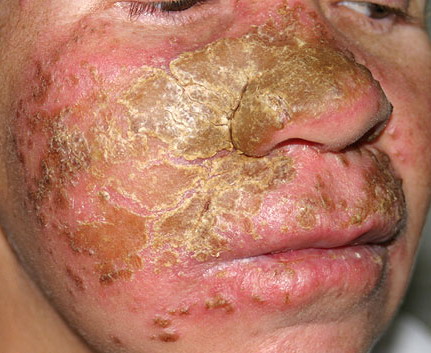
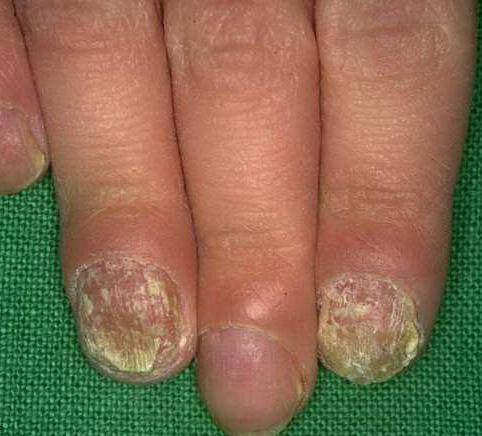
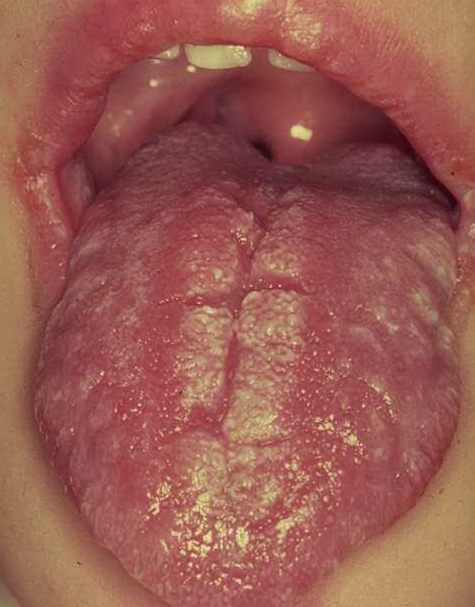
Patients with CMC have recurrent, progressive infections of the skin, nails, and mucous membranes most commonly due to C. albicans. Depending on the sub-type, the clinical presentation ranges from recurrent, recalcitrant thrush to mild erythematous scaling plaques with a few dystrophic nails to severe generalized, crusted granulomatous plaques . The cutaneous plaques occur most commonly in intertriginous areas, periorificial sites, and the scalp, but they may be generalized. The nails are thickened, brittle, and discolored, and the paronychial areas are often erythematous, swollen, and tender. Scalp infections may lead to scarring and alopecia. Although the oral mucosa is the most frequent site of mucosal alteration, esophageal, genital, and laryngeal mucosae may be affected. Strictures may be formed by candidal infection at these mucosal sites. Scrapings and cultures from cutaneous or mucosal lesions demonstrate candidal organisms.
Patients with CMC rarely develop systemic candidiasis, but 50 percent may develop recurrent or severe infections due to other organisms. In one study, 81 percent of patients with early-onset CMC also had infections with bacteria, fungi, and parasites, including bacterial septicemia. Concomitant dermatophyte infections may occur. In patients with APECED, the candidal infections tend to begin by 5 years of age, although the endocrinologic dysfunction may not be apparent until 12 to 13 years of age. The most commonly associated endocrinopathies are hypoparathyroidism (88 percent) and hypoadrenocorticism (60 percent). One-third of patients have candidiasis, hypoparathyroidism, and defective adrenal function. Other associated endocrinopathies or autoimmune disorders include gonadal insufficiency (45 percent), alopecia areata (20 percent), pernicious anemia (16 percent), thyroid abnormalities (12 percent), chronic active hepatitis or juvenile cirrhosis (9 percent), vitiligo, diabetes mellitus, and hypopituitarism. Chronic diarrhea and malabsorption have been reported in 25 percent of patients and usually are associated with hypoparathyroidism. Some affected patients also have pulmonary fibrosis, dental enamel hypoplasia, and keratoconjunctivitis. The “ectodermal dysplasia” features are likely to be secondary to the candidal infections or autoimmunity44 Patients with APECED often have autoimmune antibodies, including antithyroglobulin, antimicrosomal antibodies, antiadrenal and antimelanocyte antibodies, and rheumatoid factor. Autoantibodies also have been found in patients with CMC who do not have clinical endocrinologic disease.
PROGNOSIS, CLINICAL COURSE, AND TREATMENT
Candidal lesions in patients with CMC generally respond to systemically administered azole antifungal agents (itraconazole, fluconazole) or terbinafine. Ketoconazole is no longer used because of the risk of hepatitis. Patients who are resistant usually respond to amphotericin B with or without flucytosine. Cutaneous granulomas often are less responsive despite clearance of infection. Recurrences are common, and the antifungal agents must be used intermittently. The drugs have no effect on the abnormal cell-mediated immunity. All patients with CMC should have an annual endocrine evaluation and patients with documented endocrinopathy or a family history of APECED should be monitored more closely. Patients who have a history of infections other than candidal should have further
|
الترجمة الفورية للصفحة |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||