JUVENILE
XANTHOGRANULOMA
Clinical Findings
CUTANEOUS LESIONS
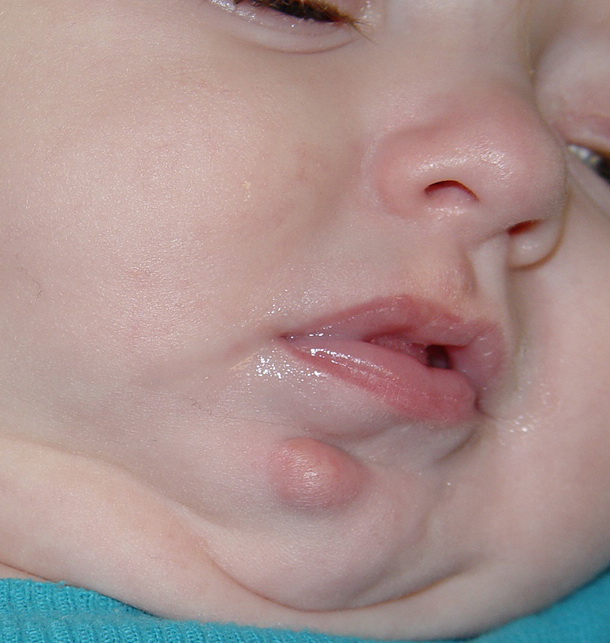
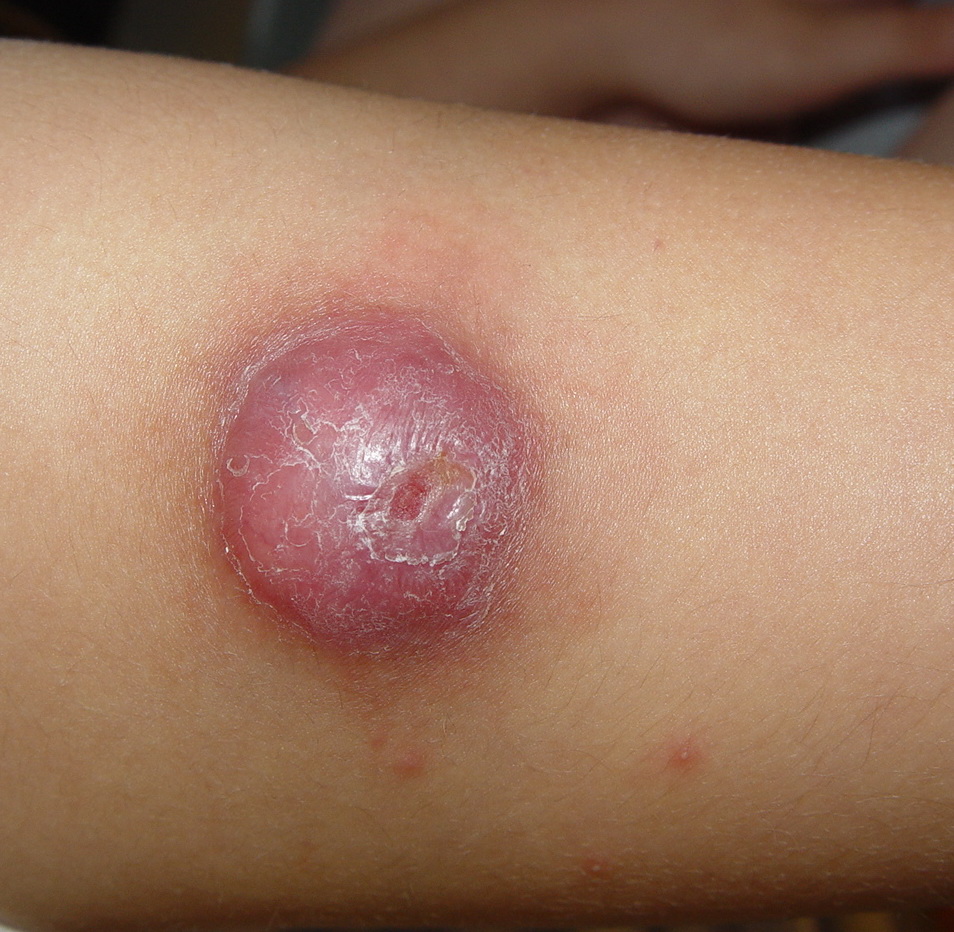
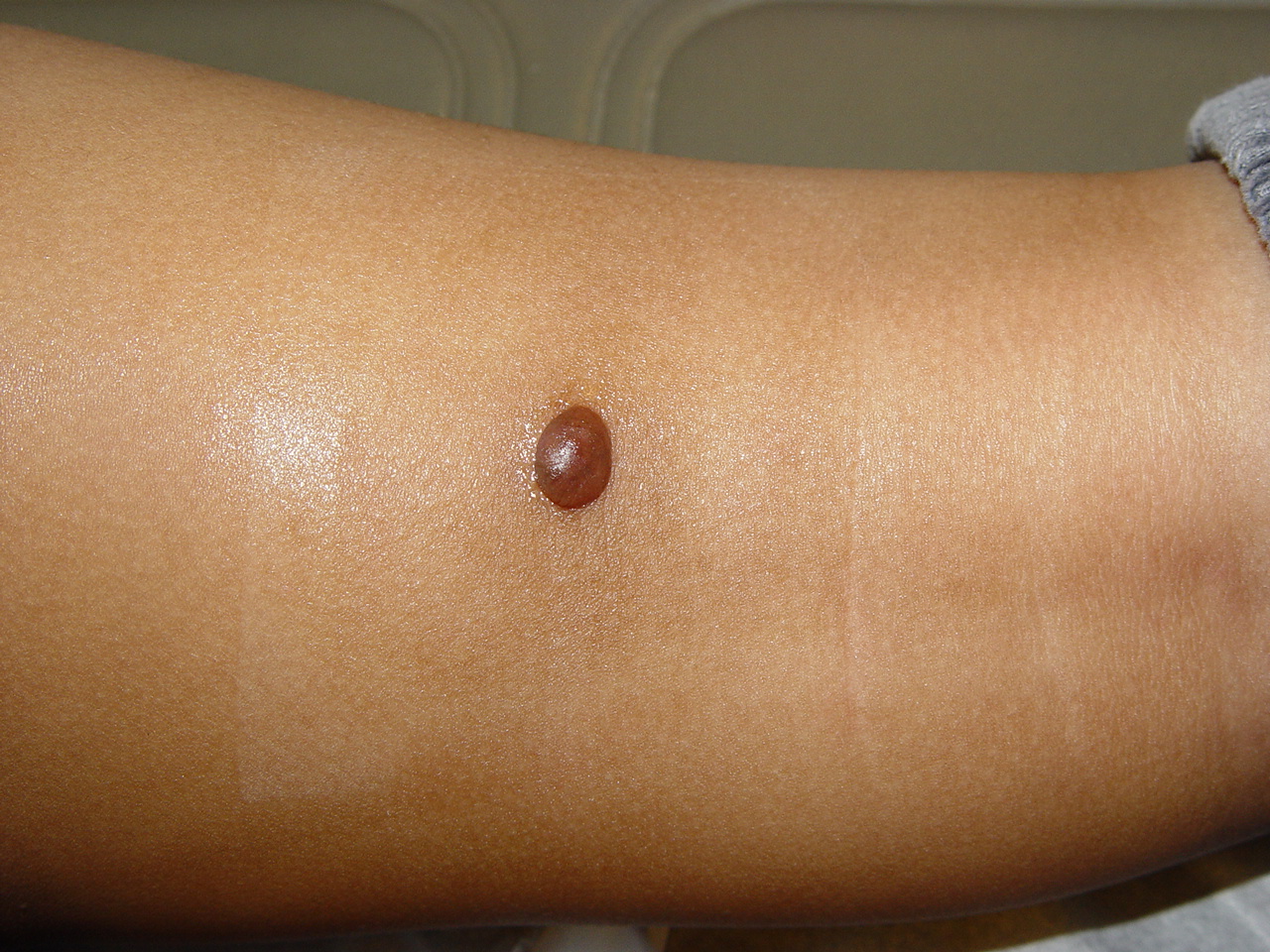
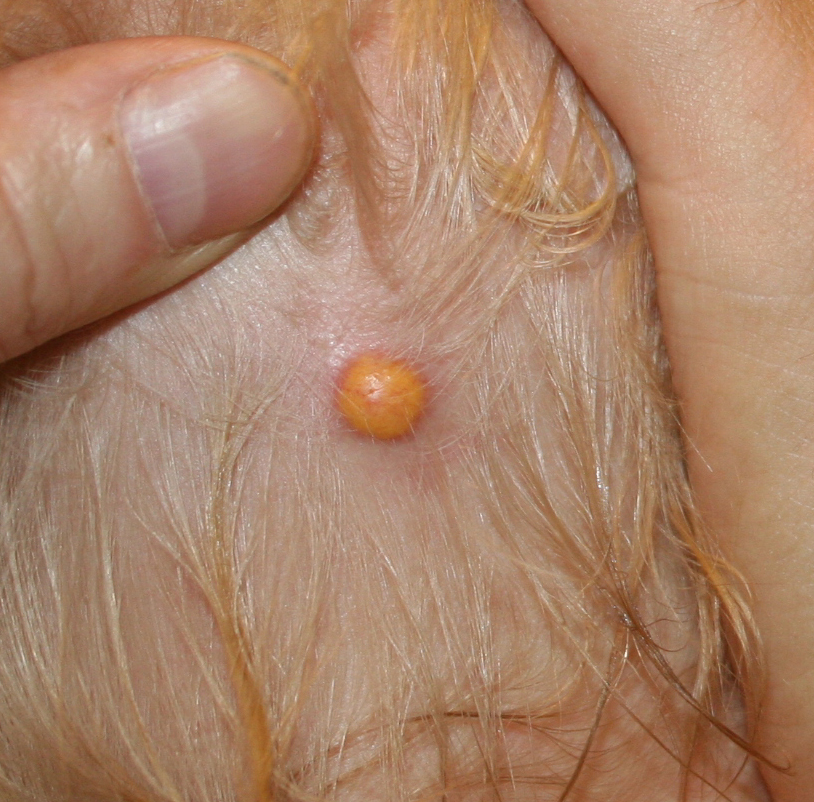
JXG is a benign, self-healing disorder that is characterized by asymptomatic yellowish papulonodular lesions of the skin and other organs in the absence of a metabolic disorder. Lesions consist of an infiltrate of histiocytes with a progressively greater degree of lipidation.
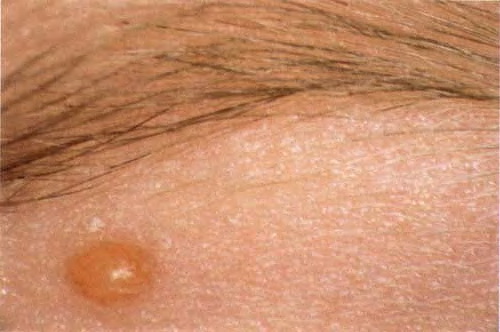
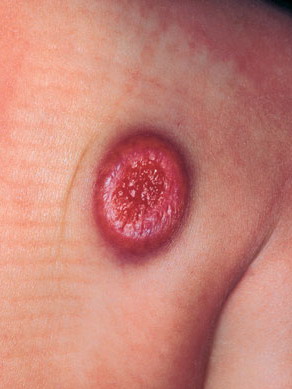
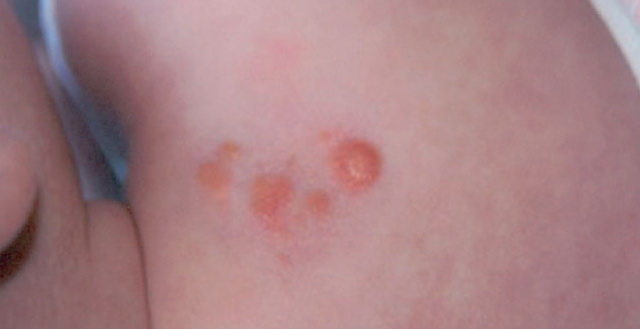
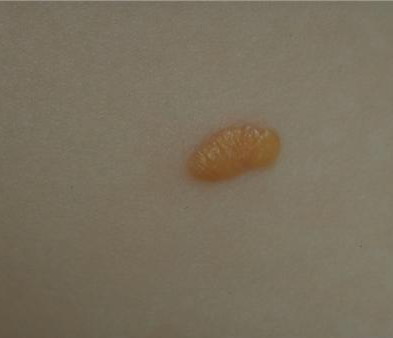
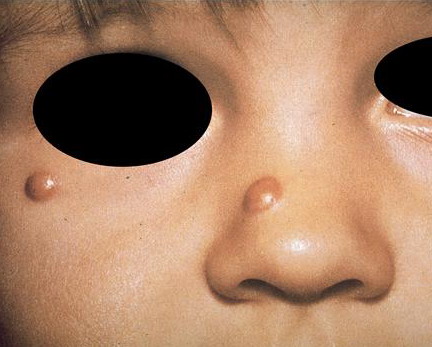
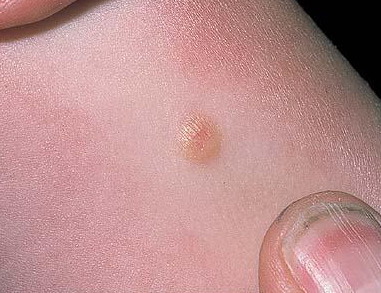
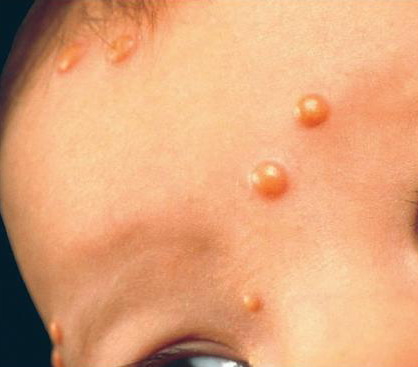
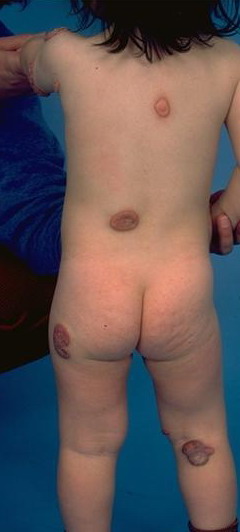
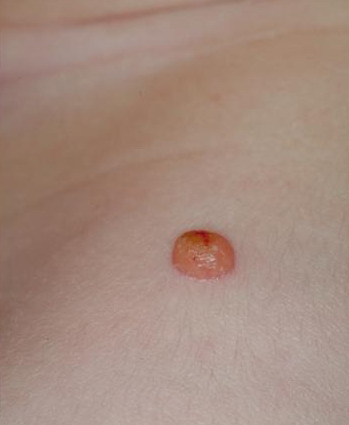
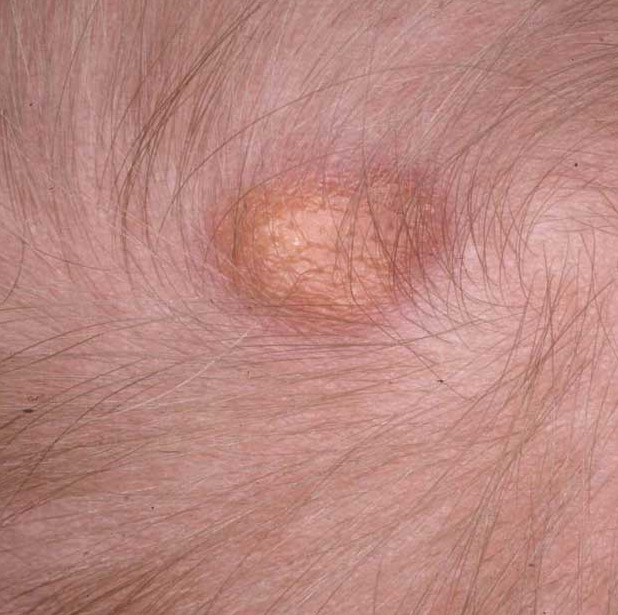
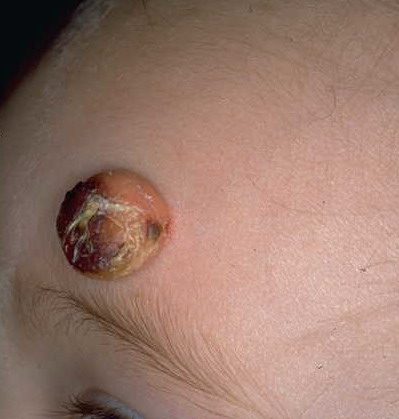
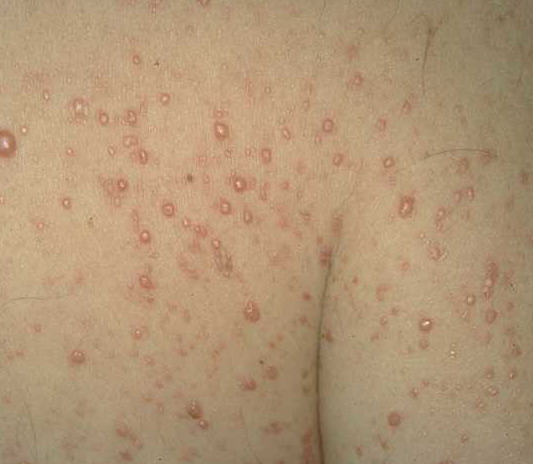
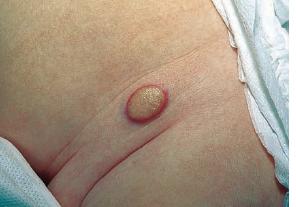
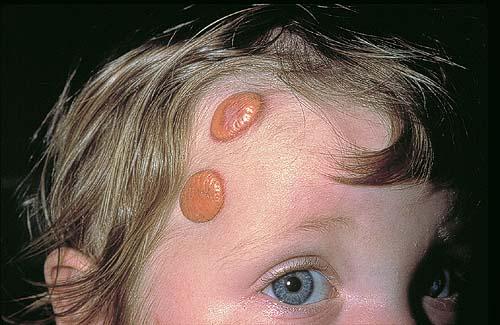
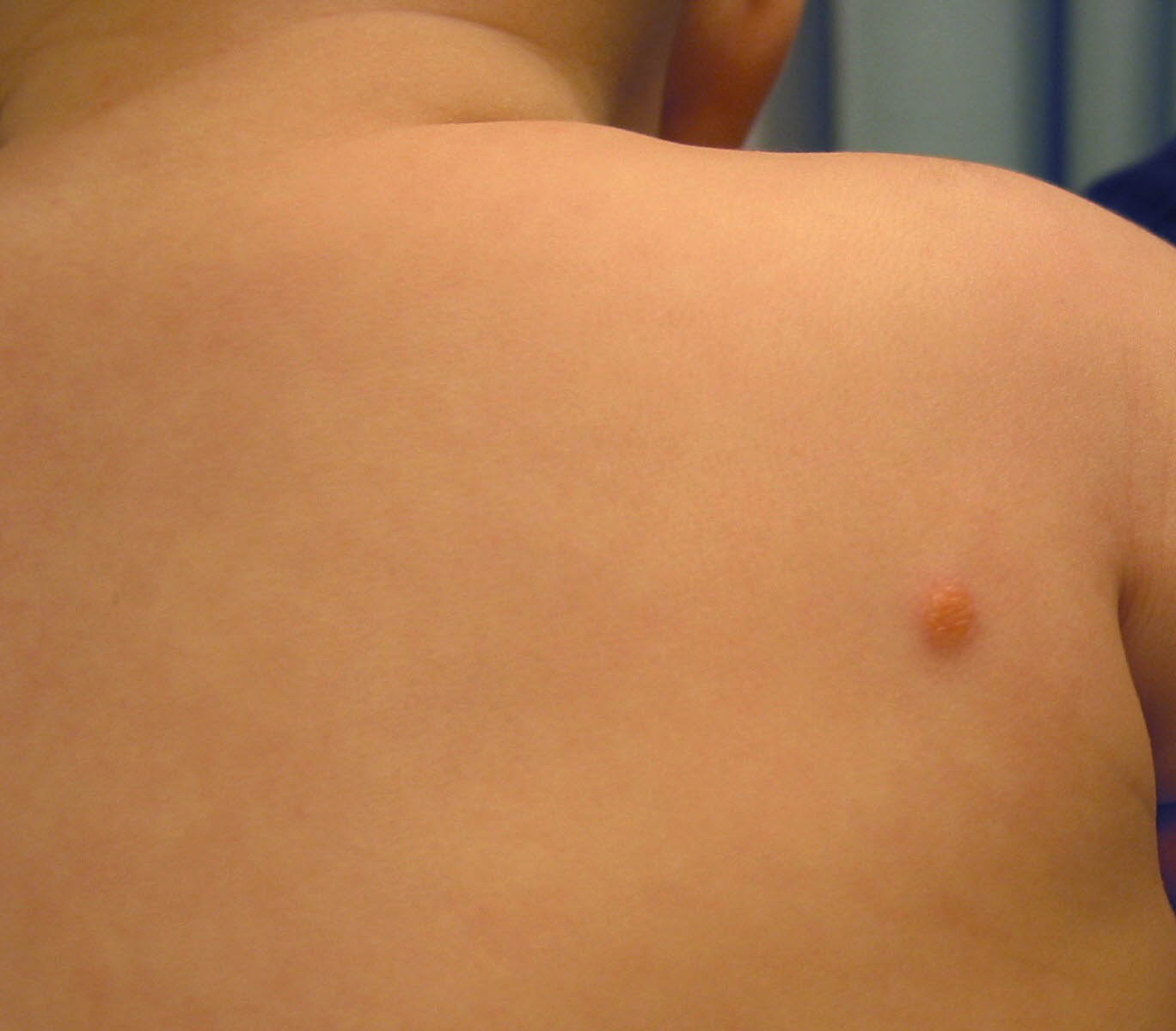
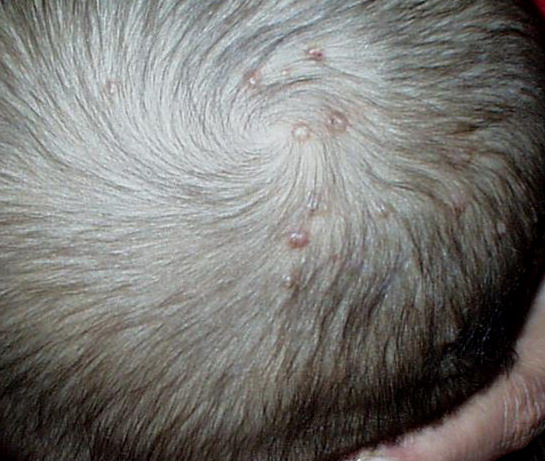
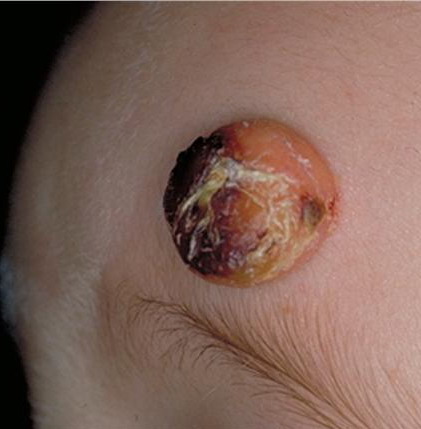
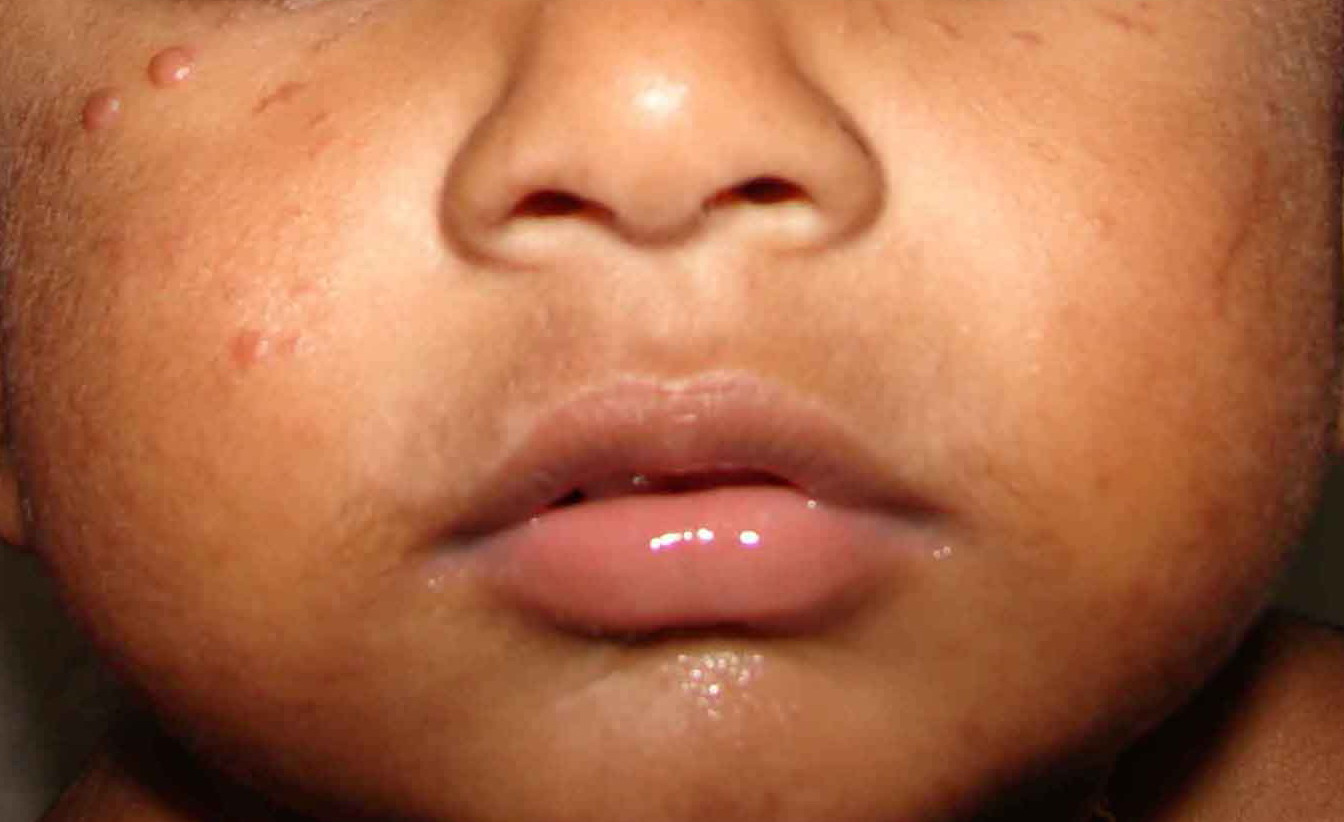
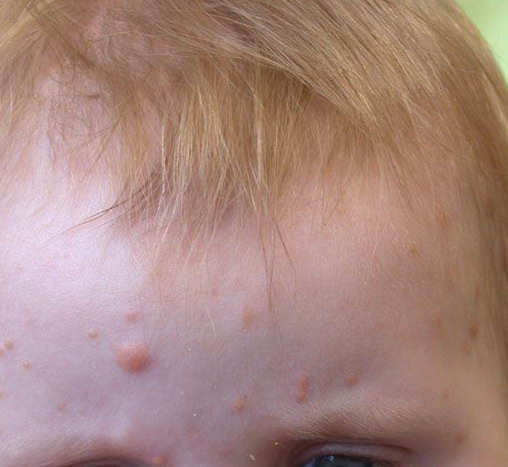
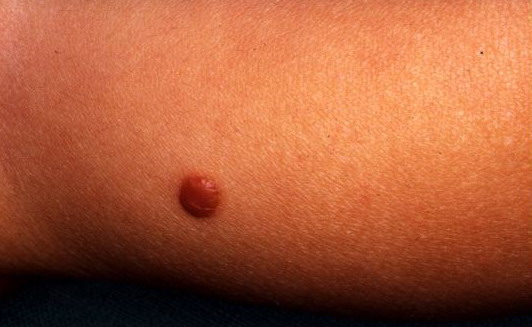
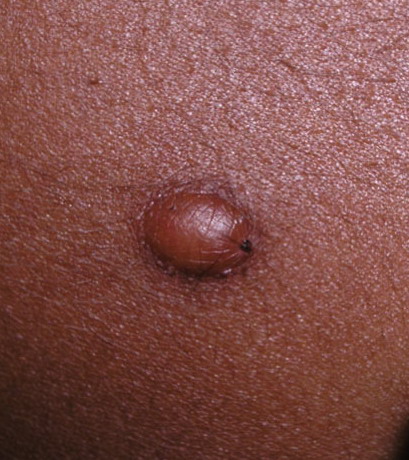
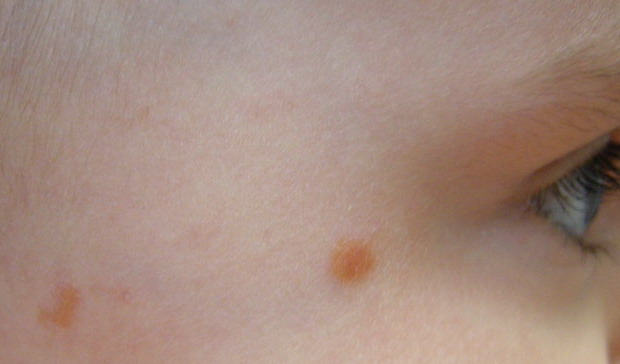
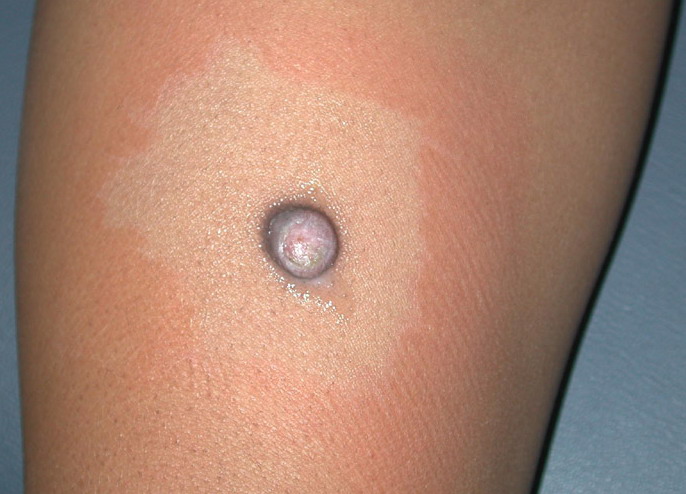
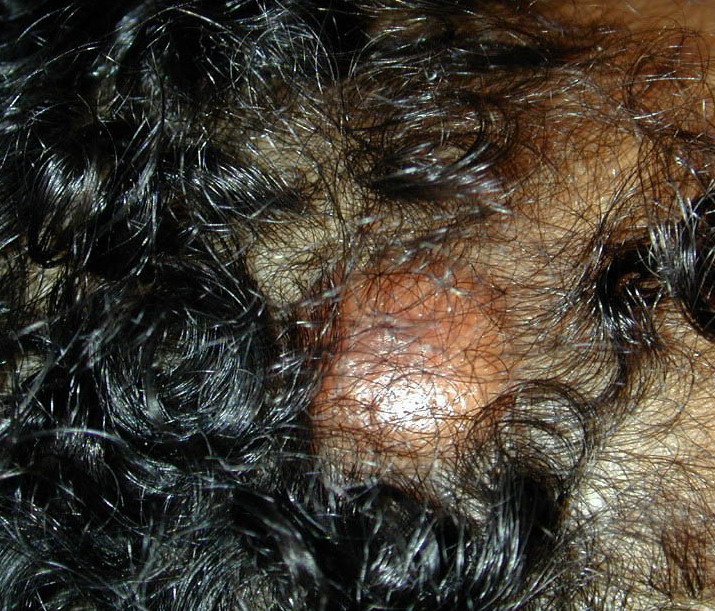
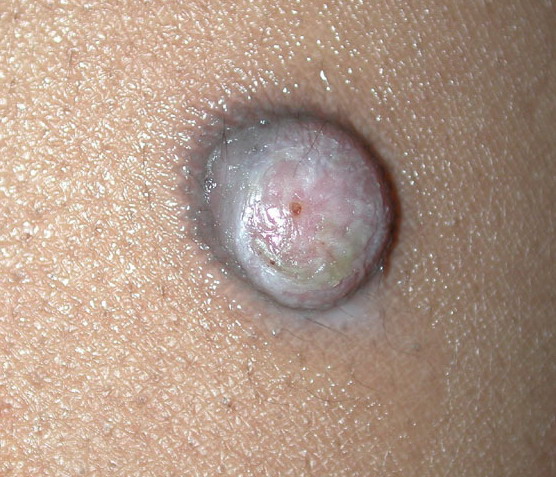
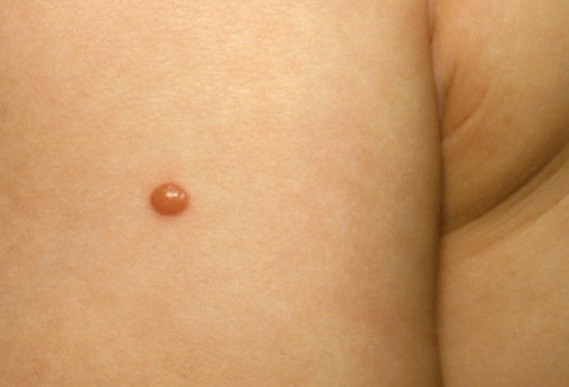
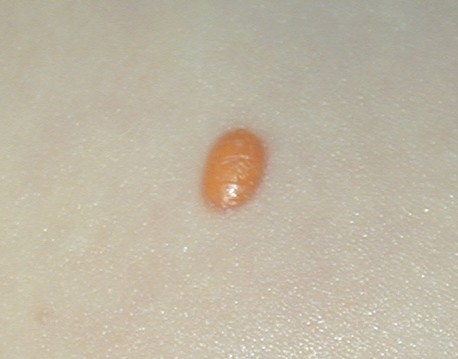
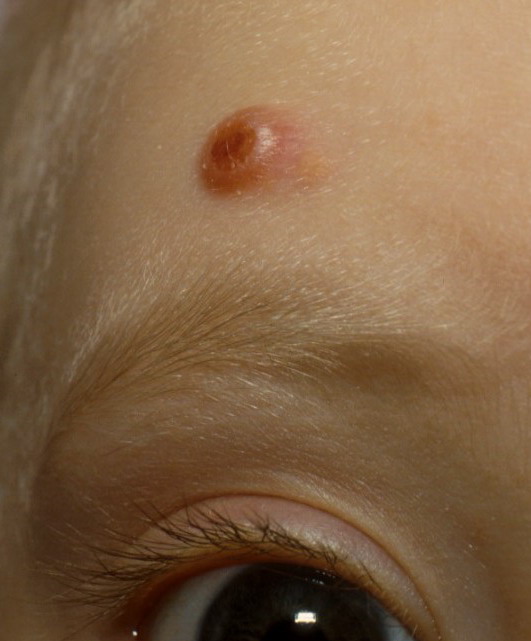
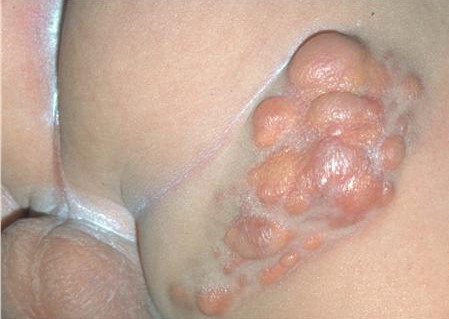
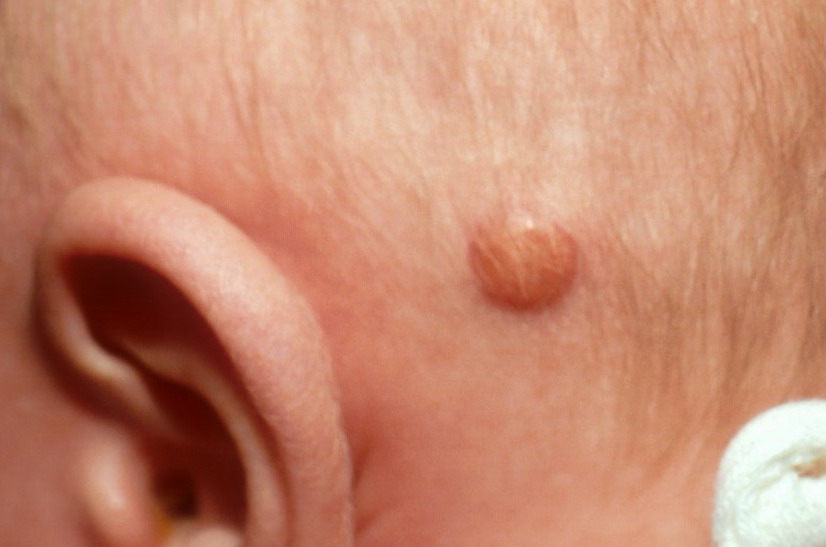
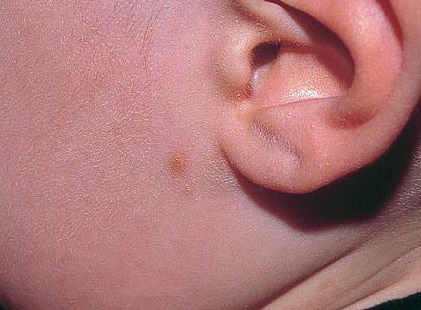
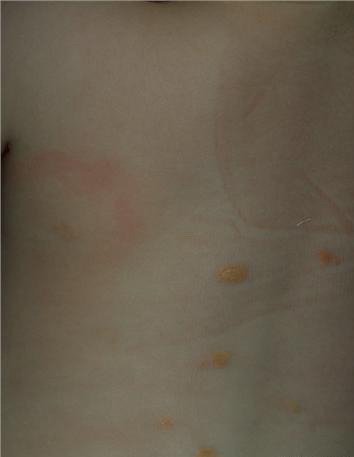
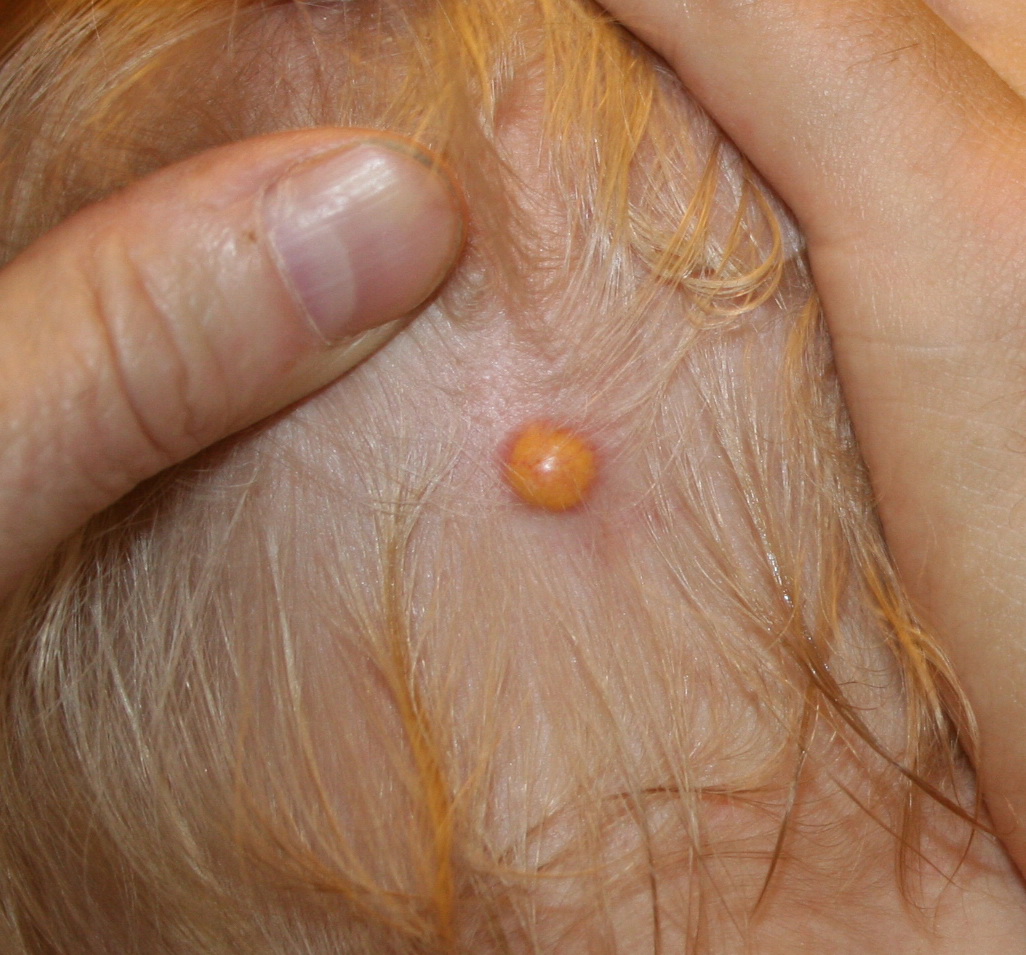
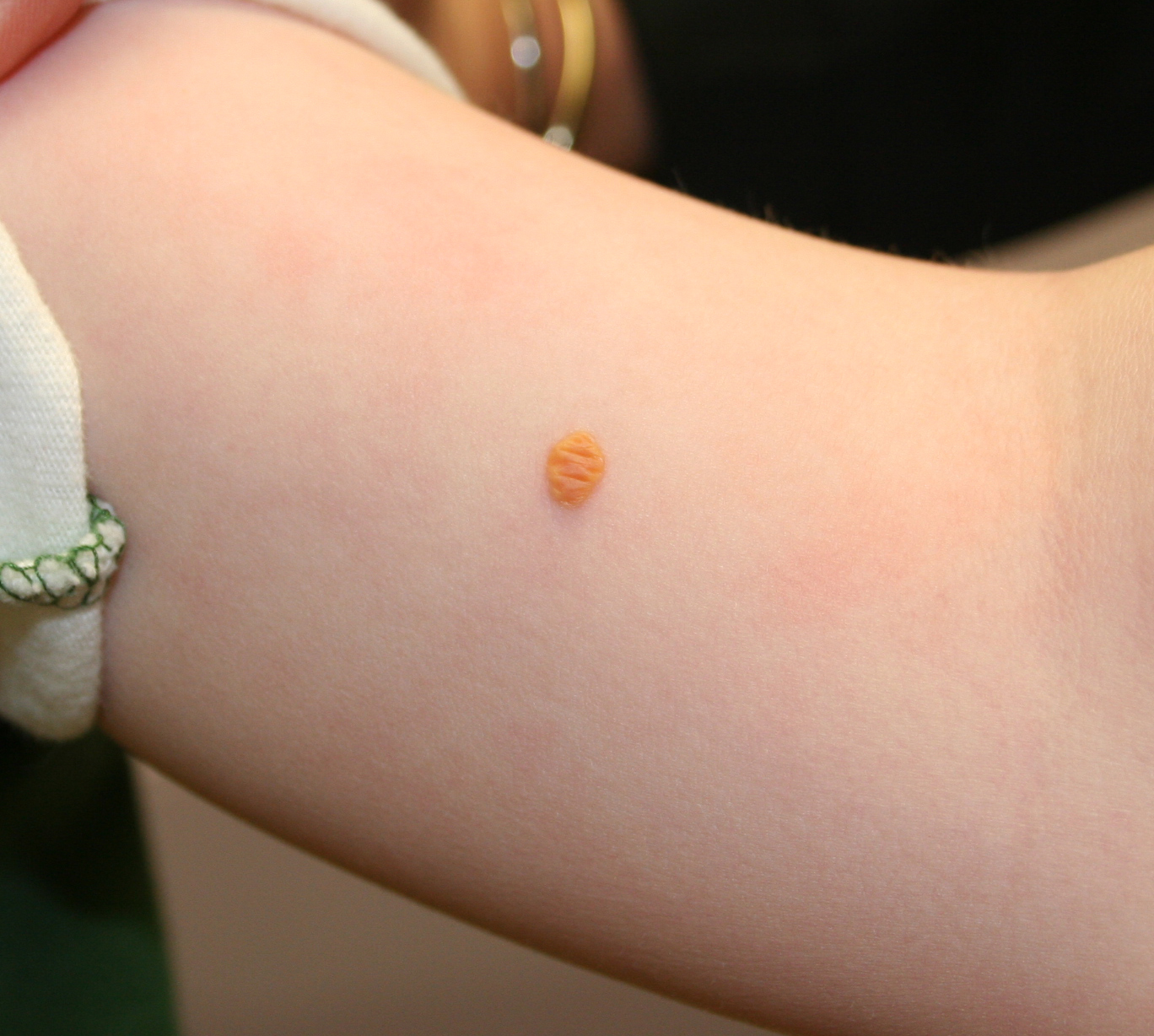
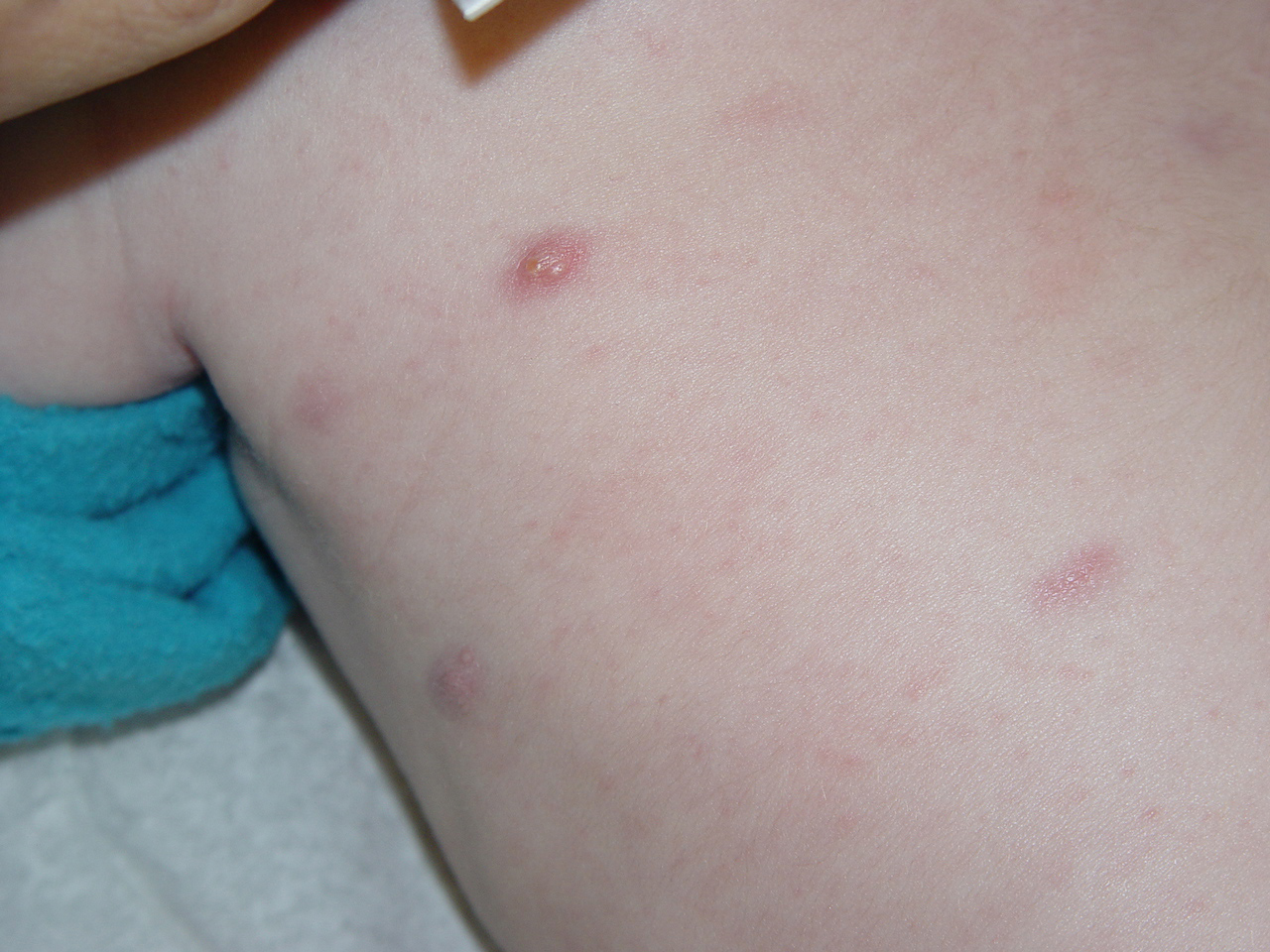
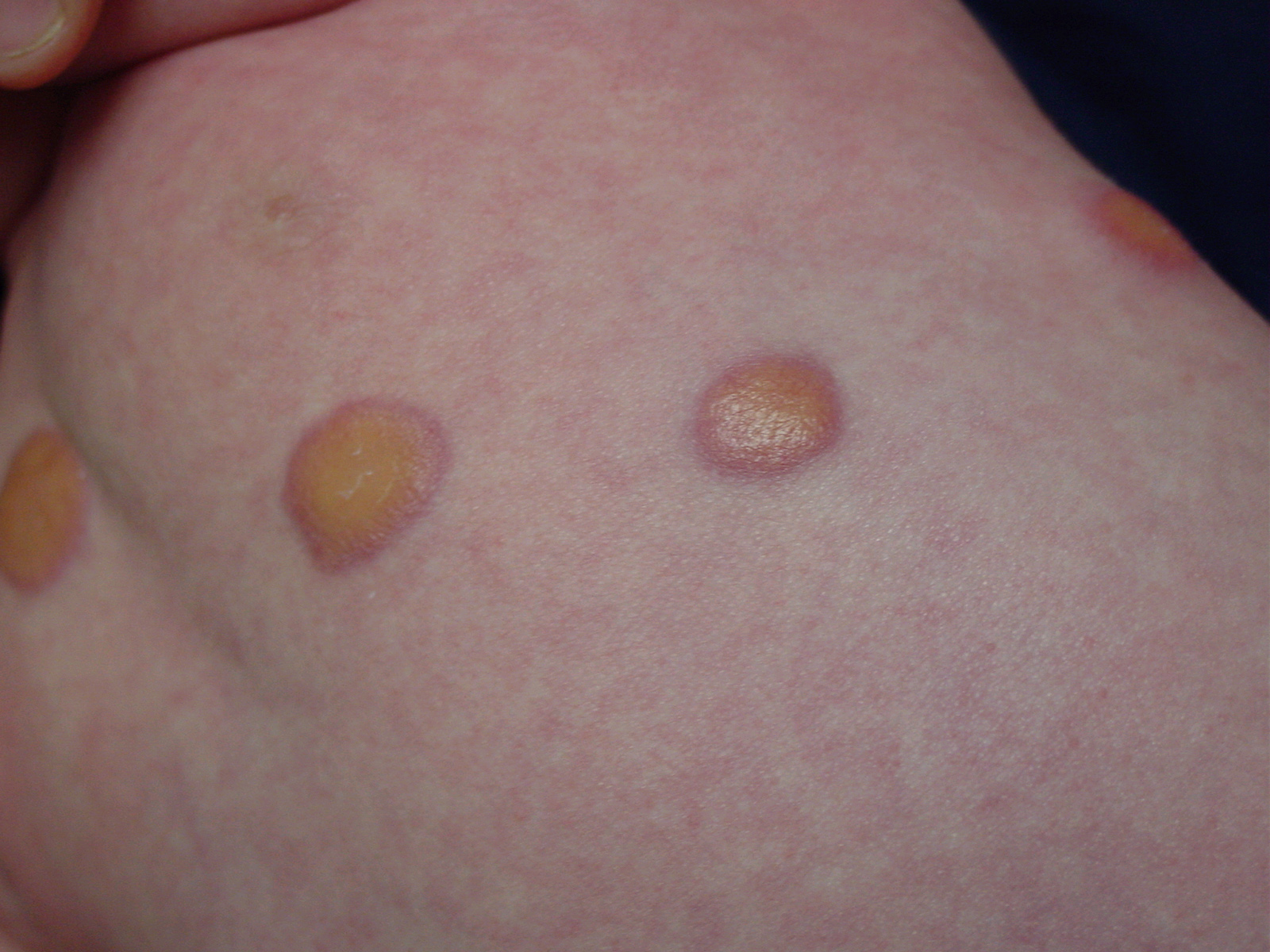
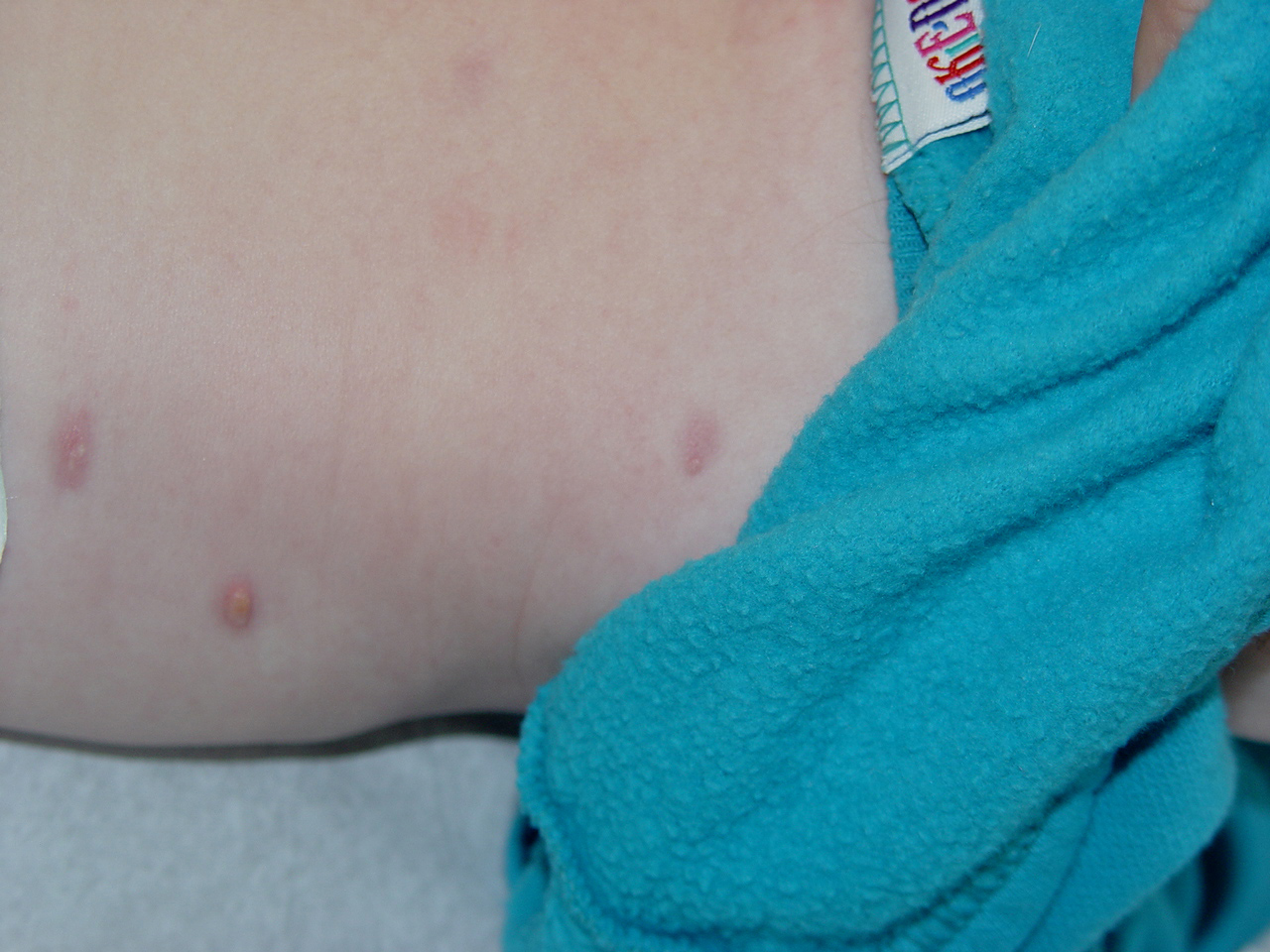
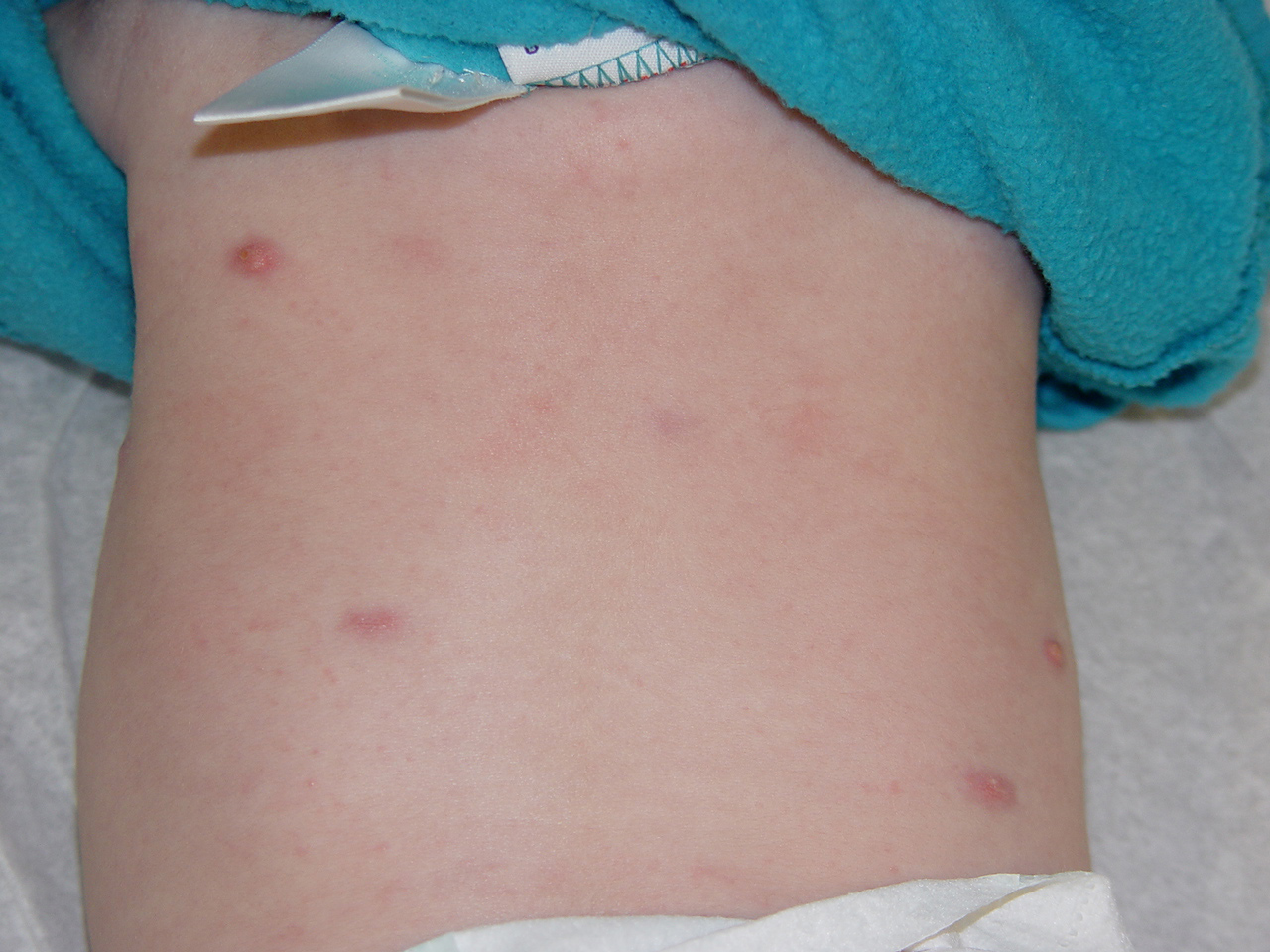
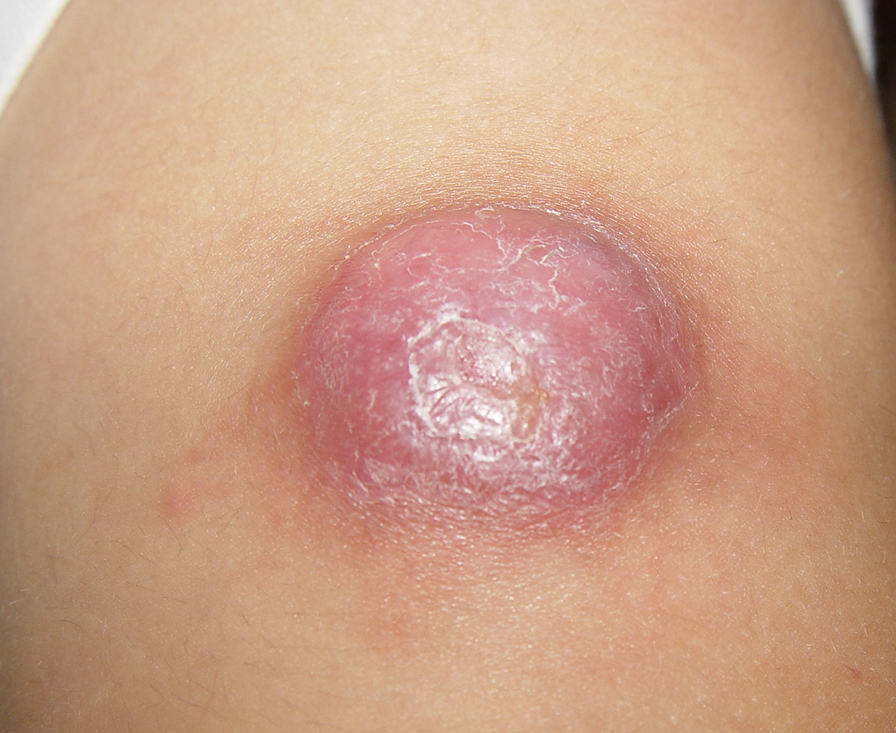
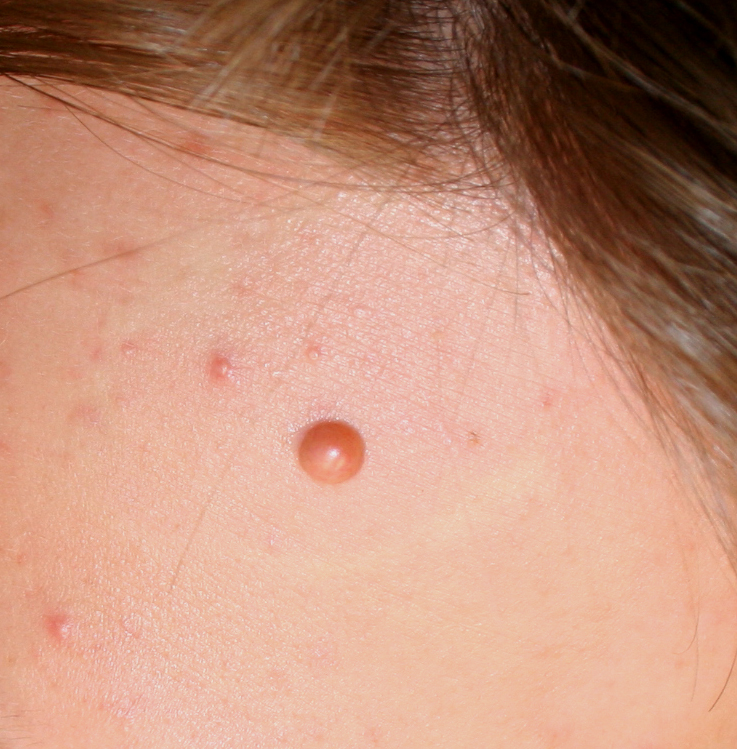
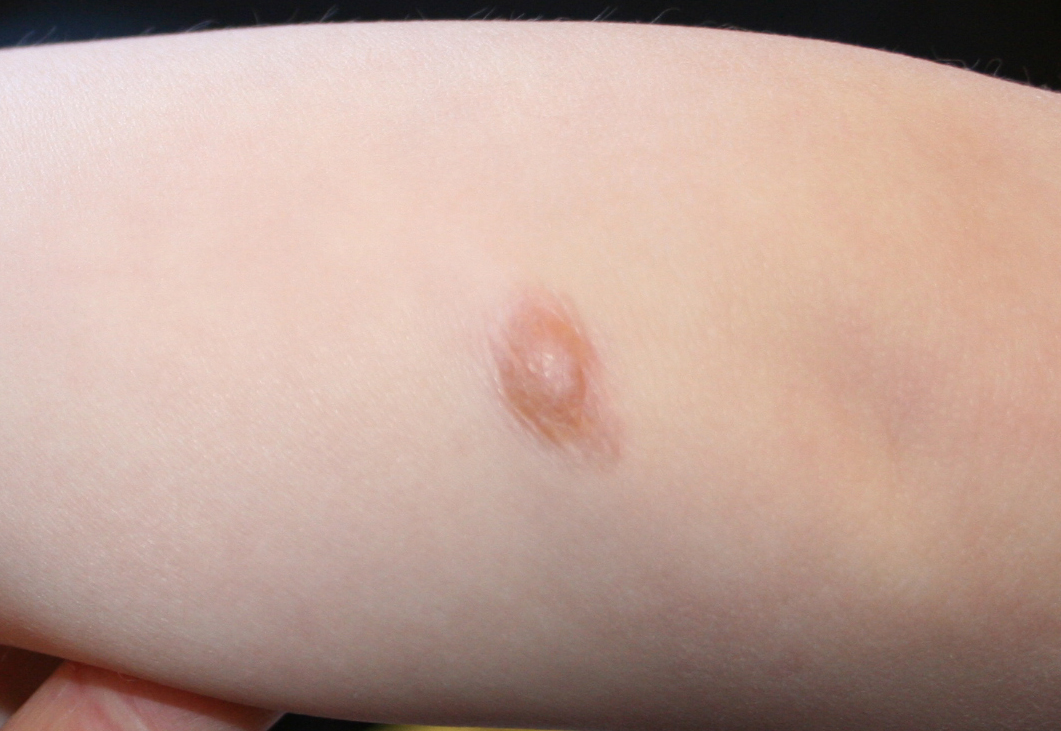
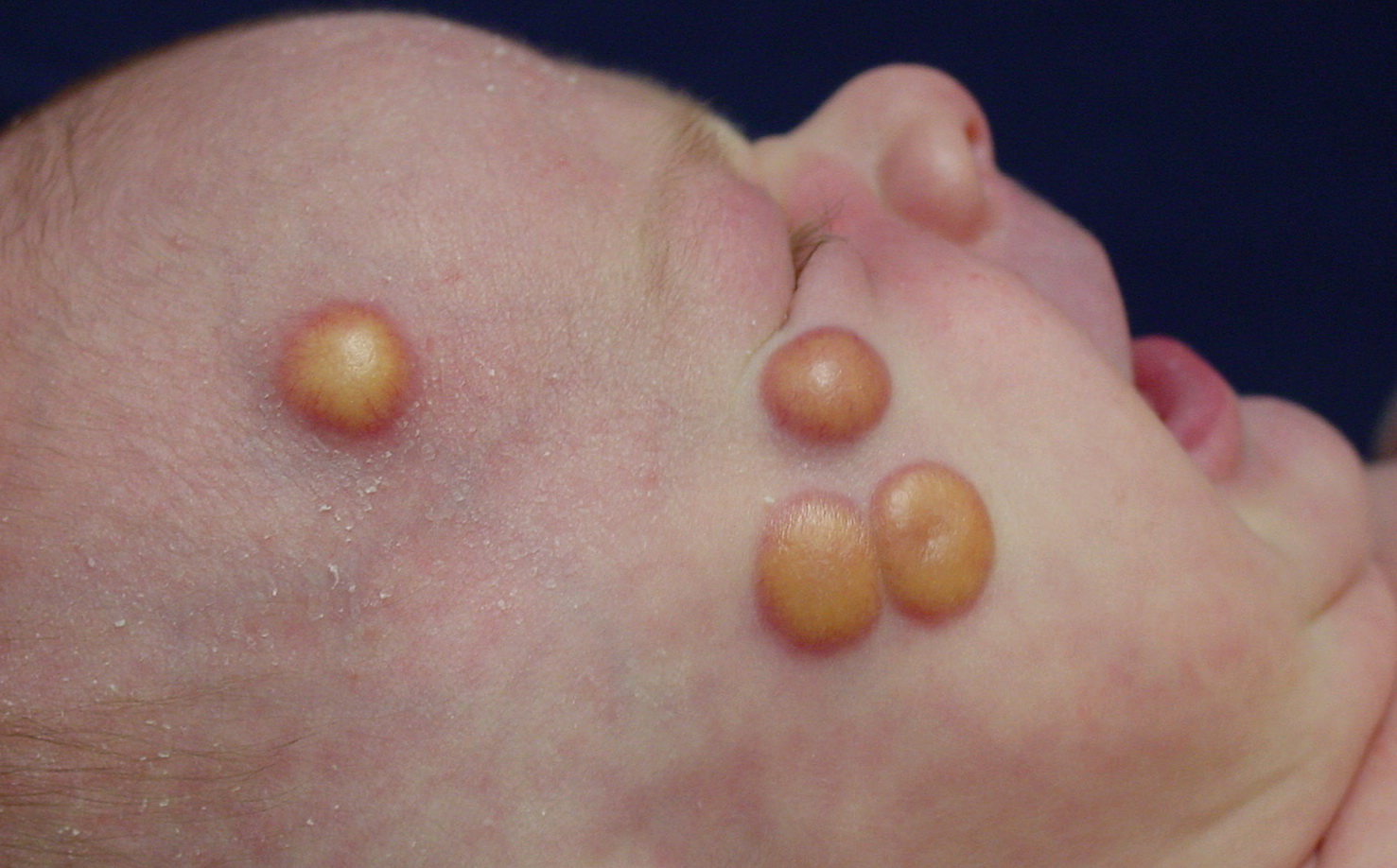
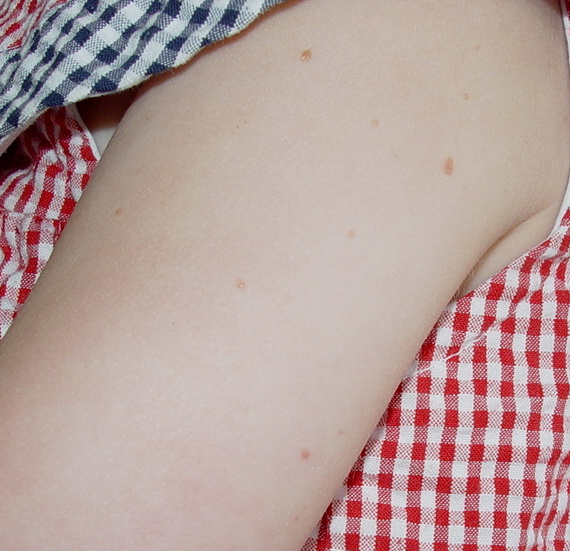
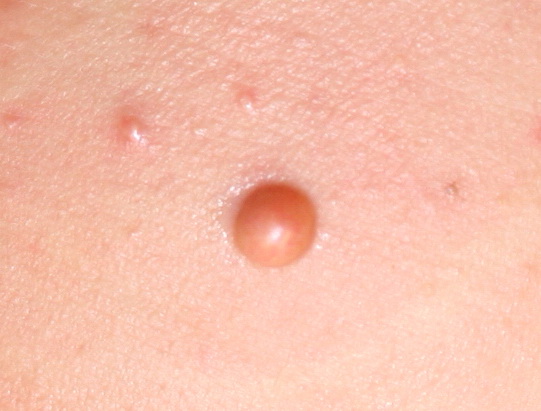
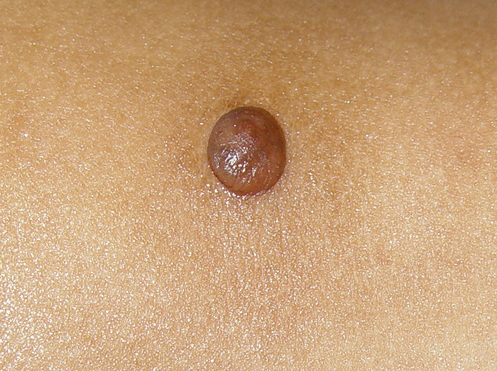
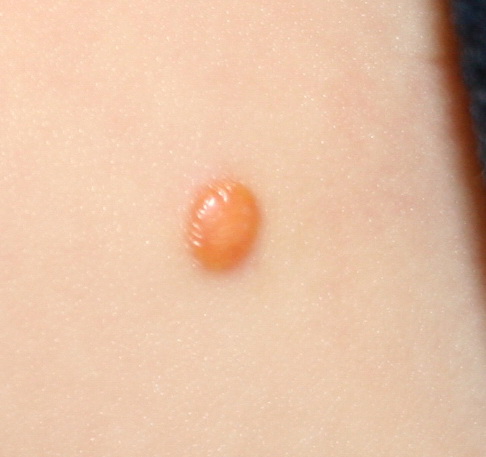
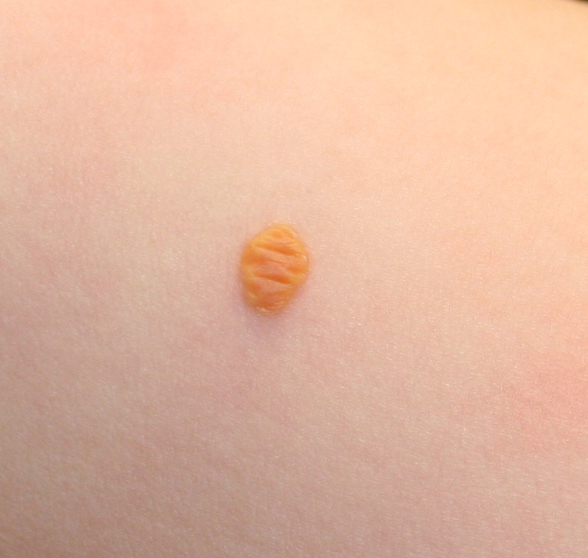
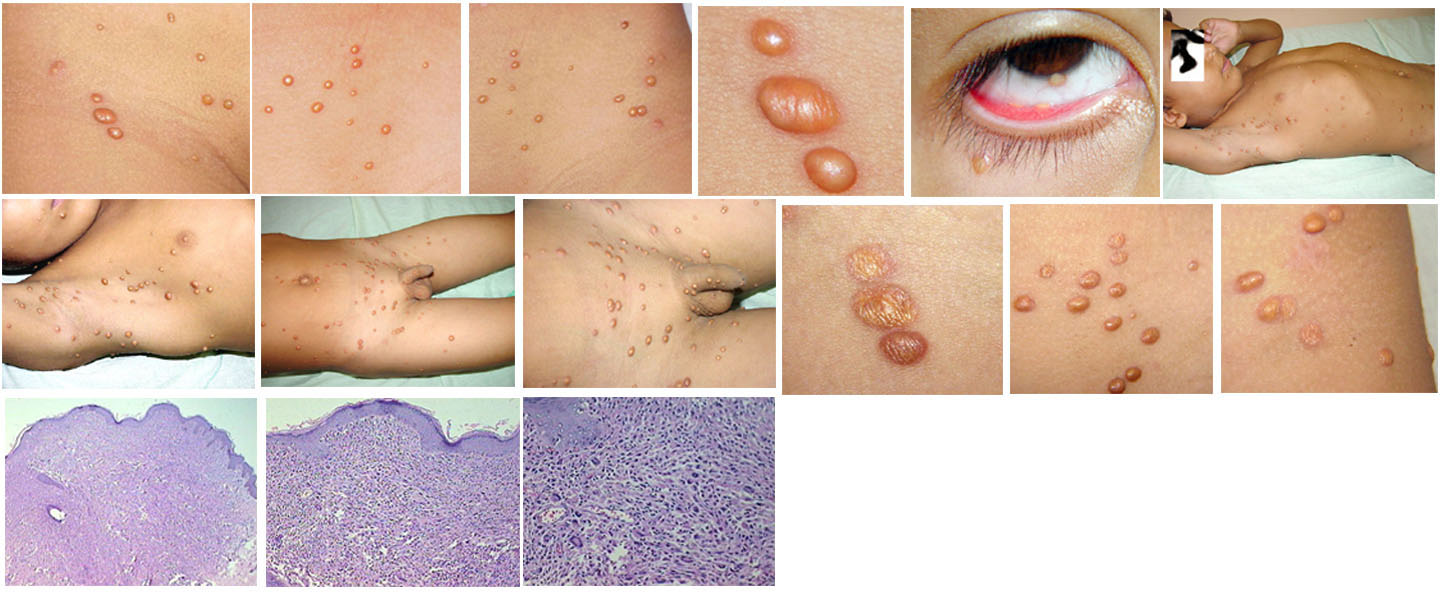
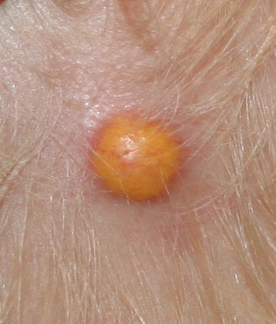
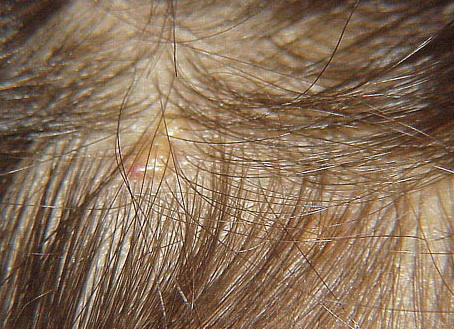
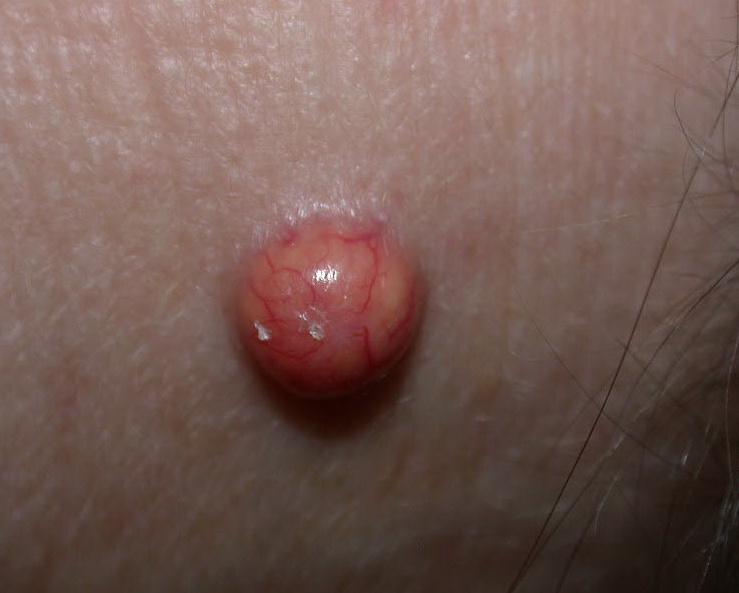
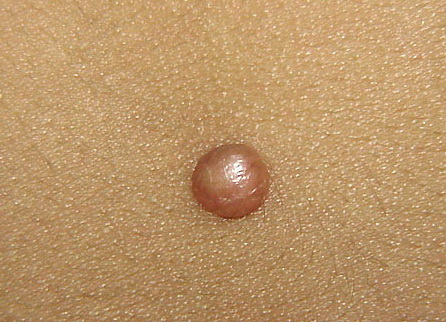
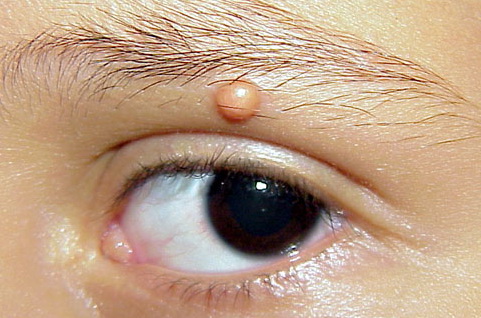
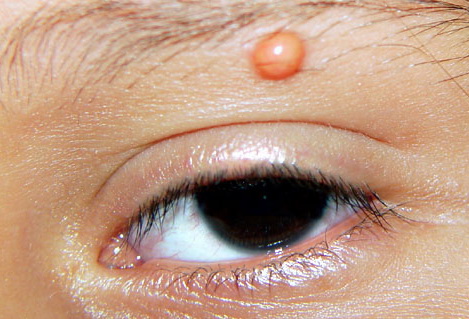
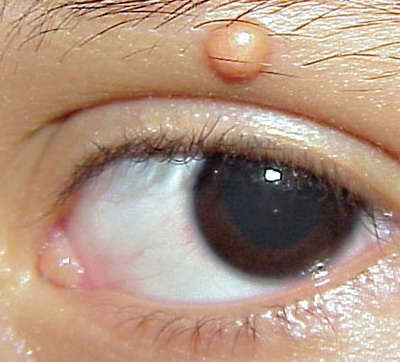
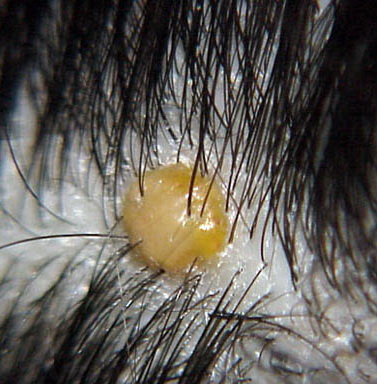
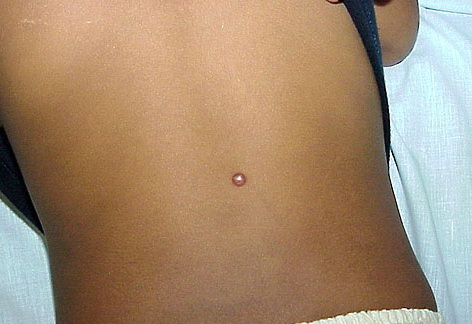
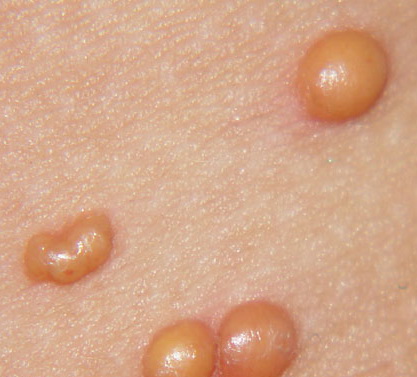
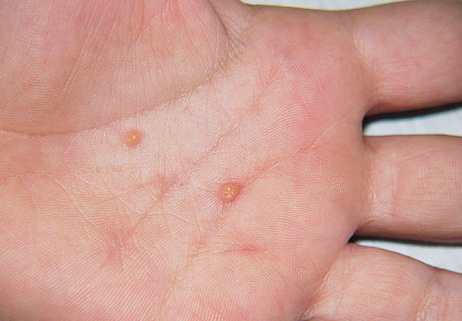
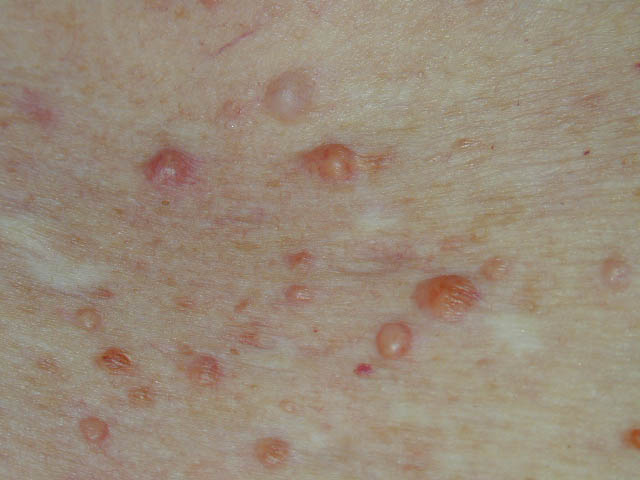
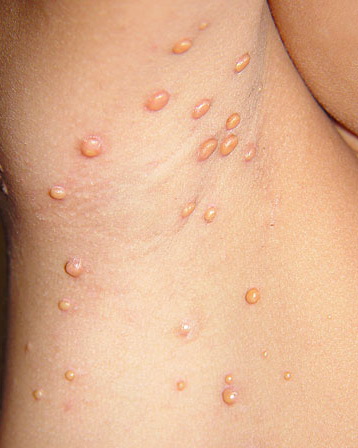
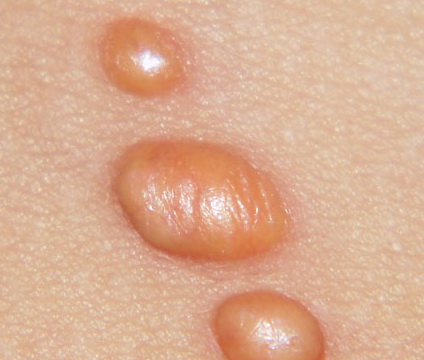
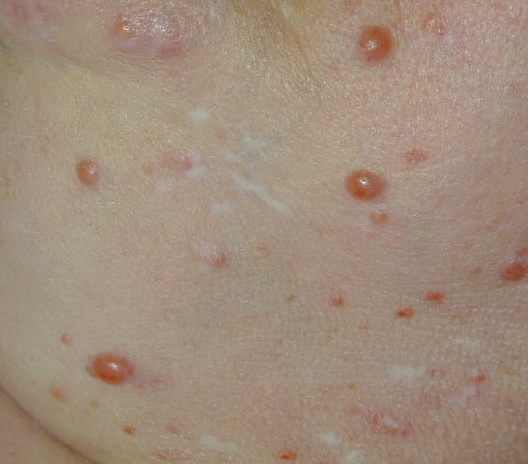
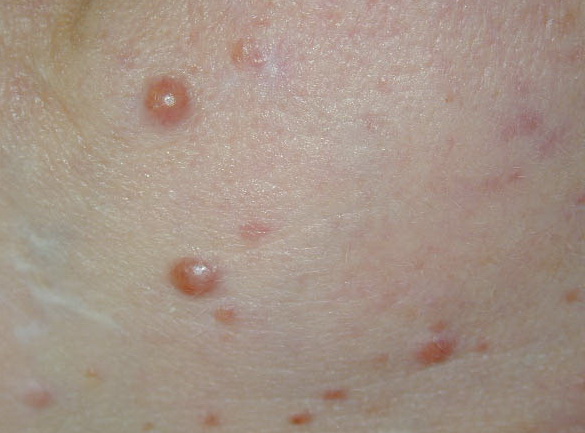
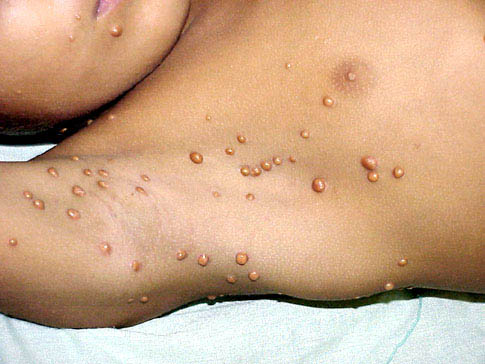
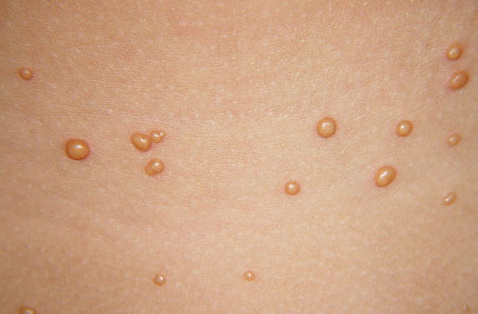
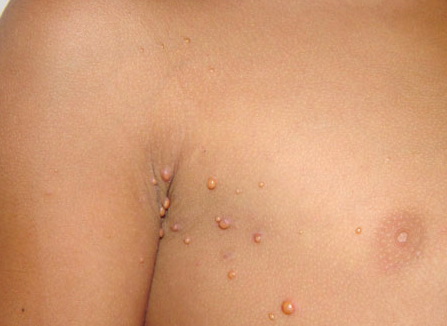
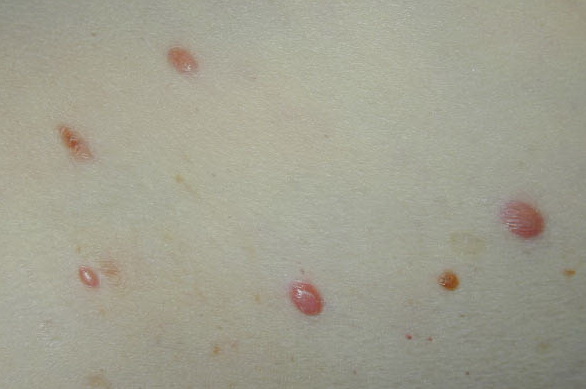
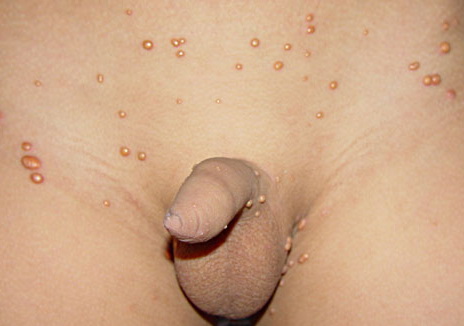
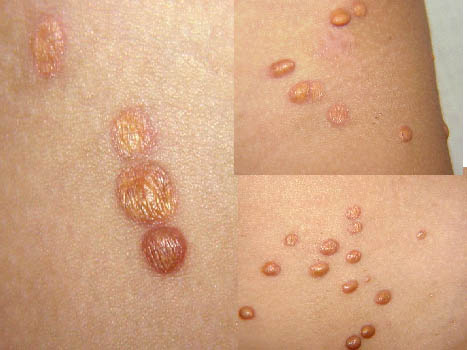
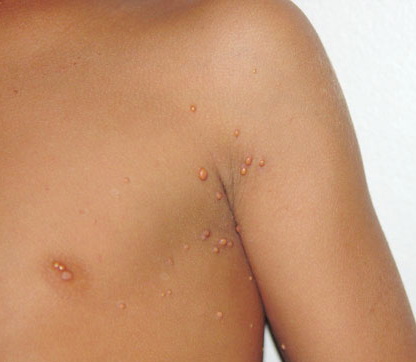
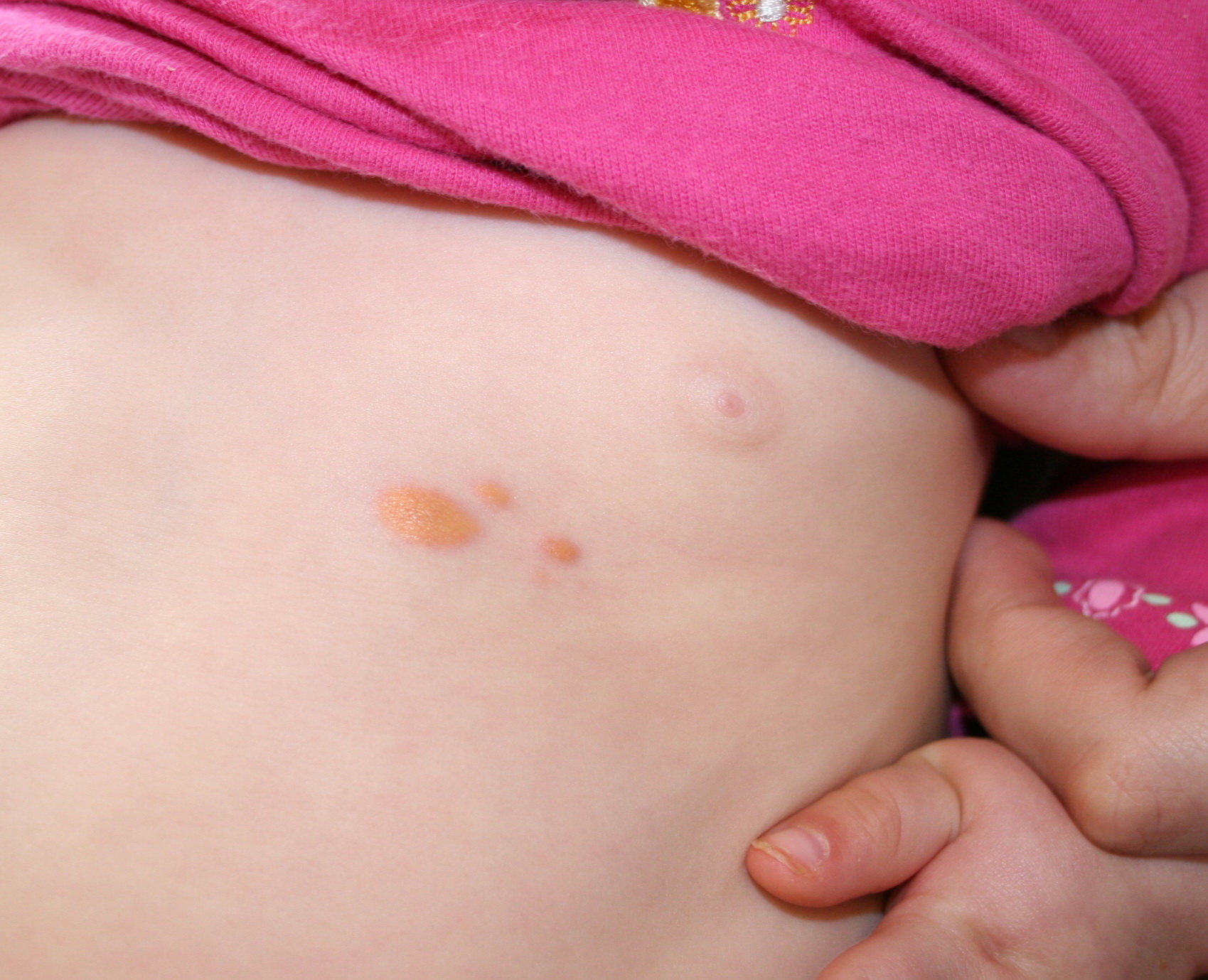
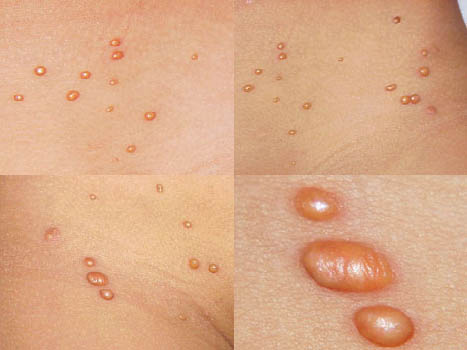
In JXG two main clinical forms can be distinguished, a papular and a nodular form. In both the lesions are initially orange-red or red-brown but quickly turn yellowish. The papular form is characterized by numerous (up to
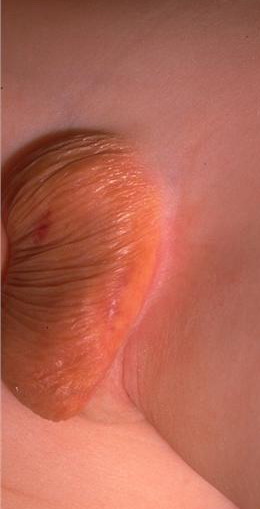
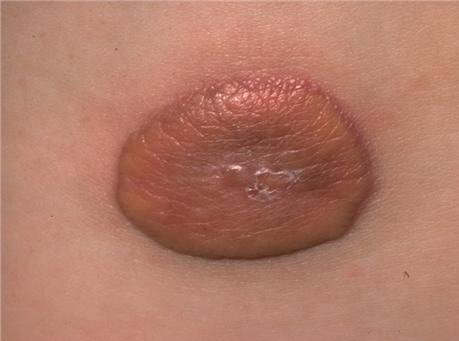
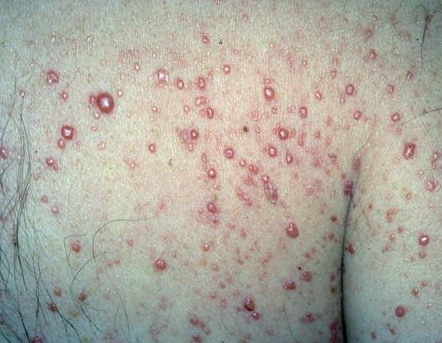
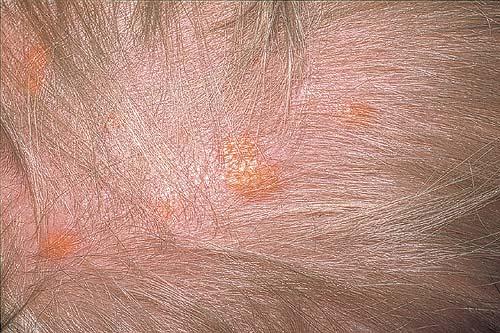
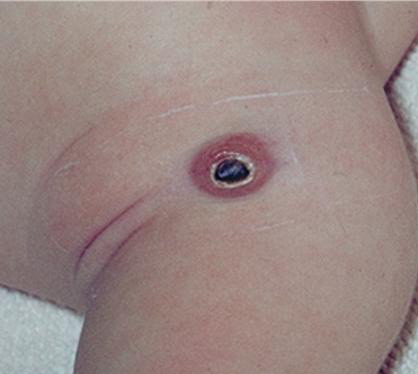
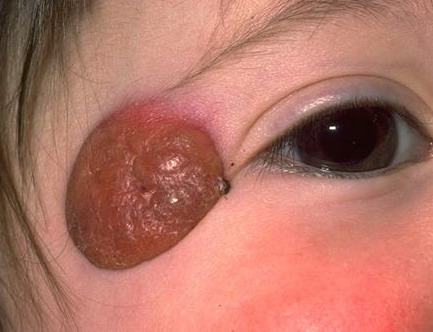
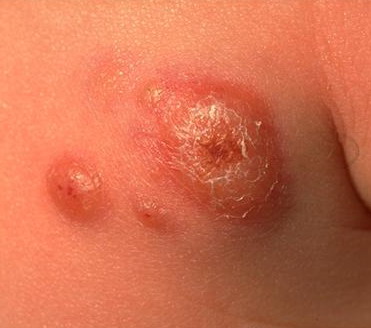
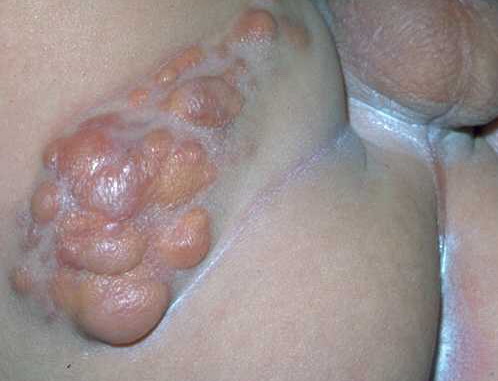
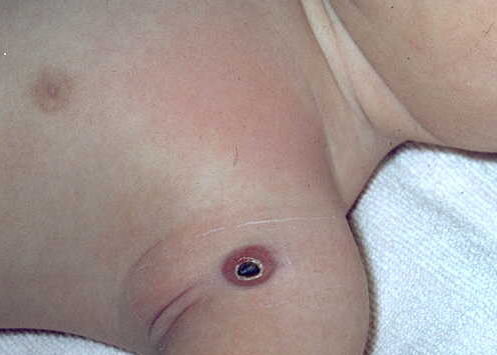
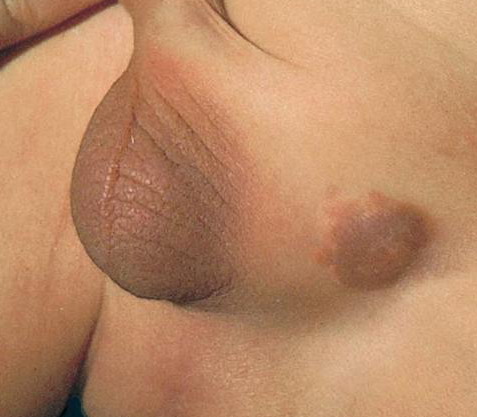
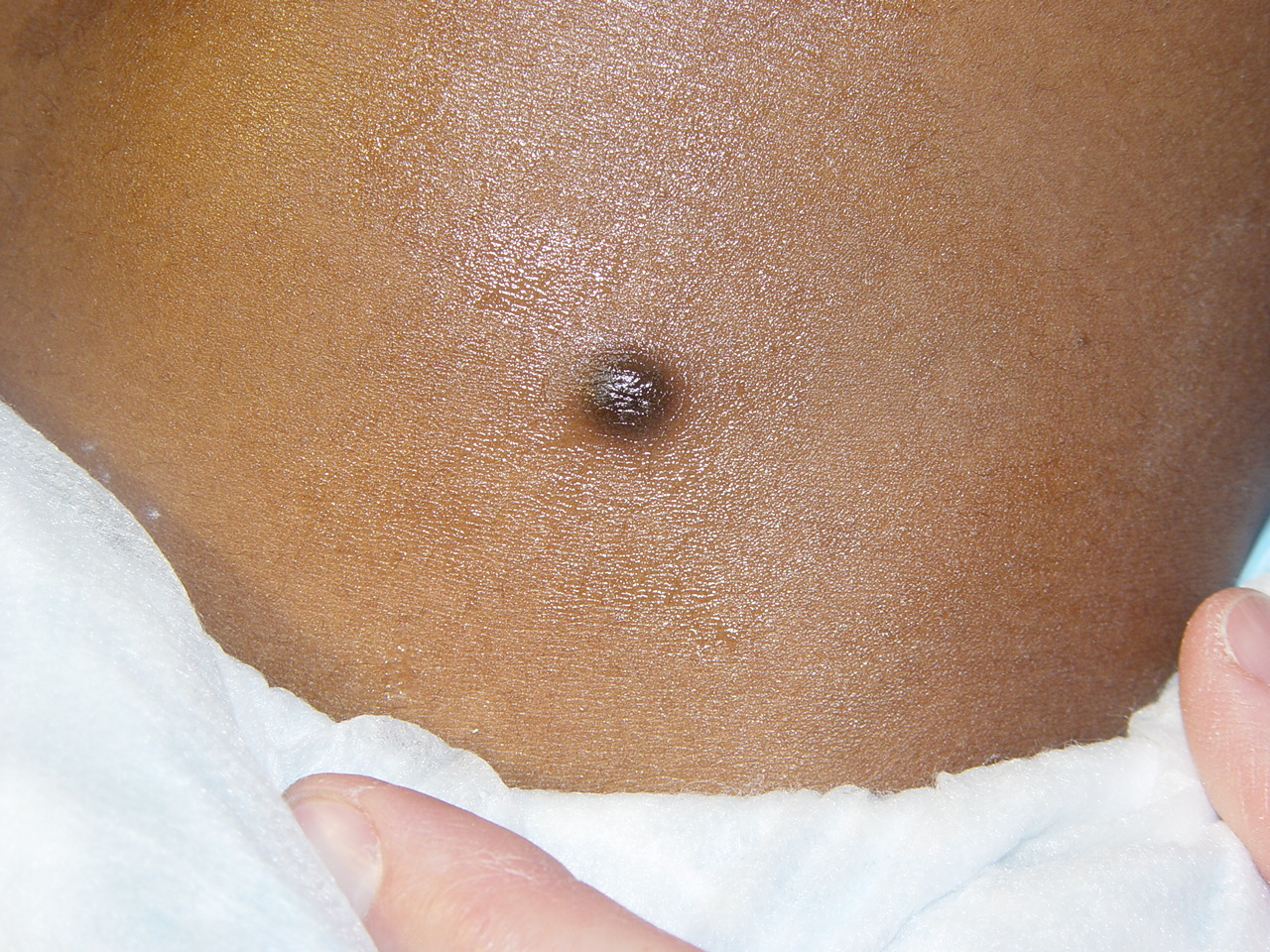
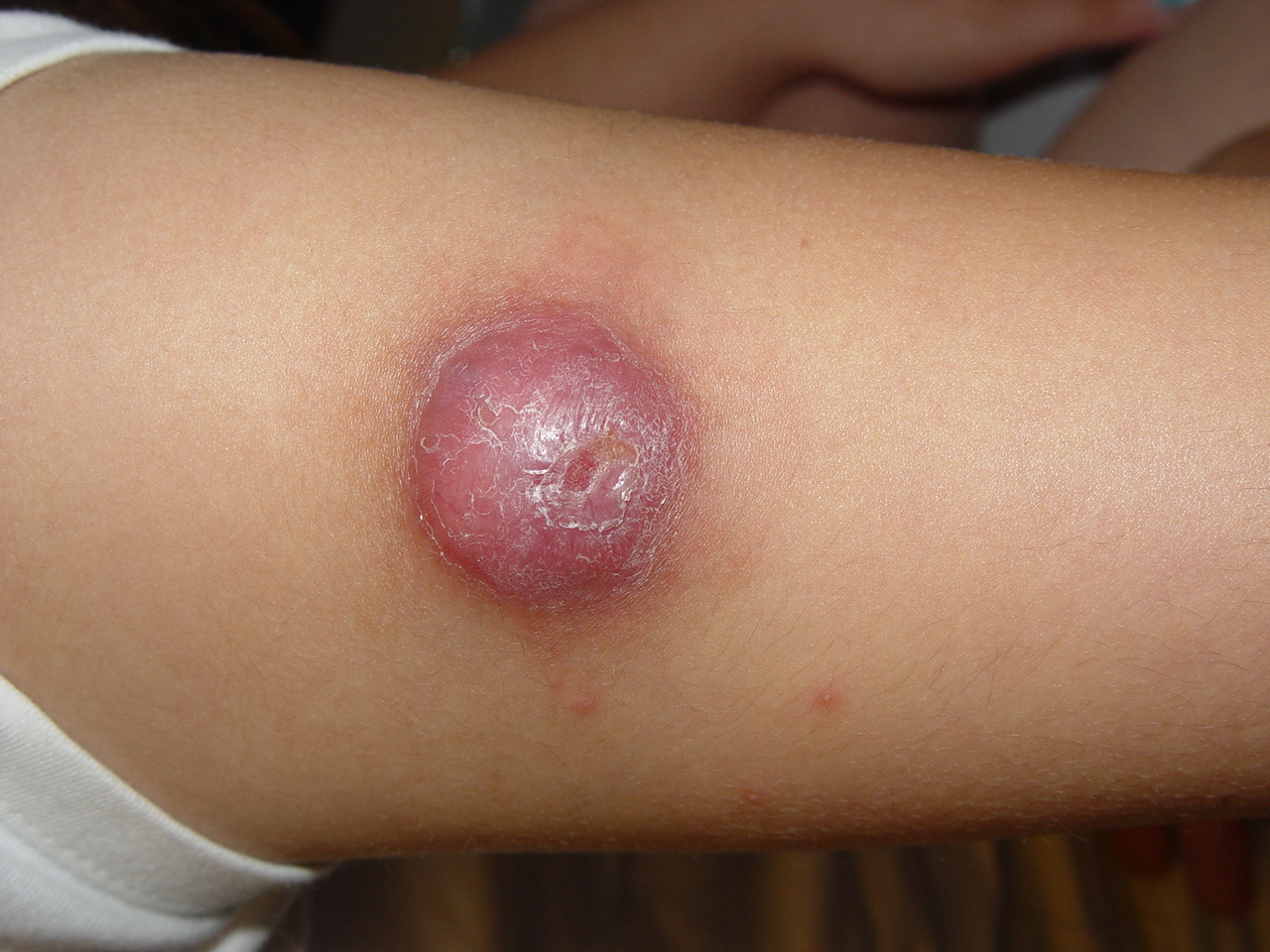
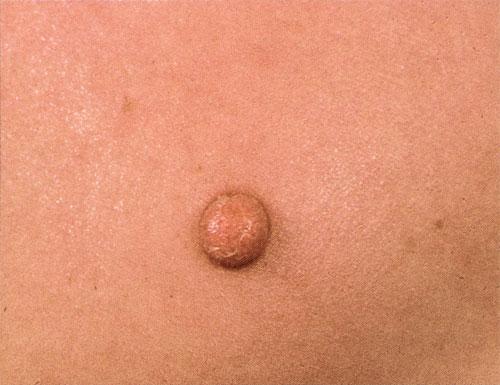
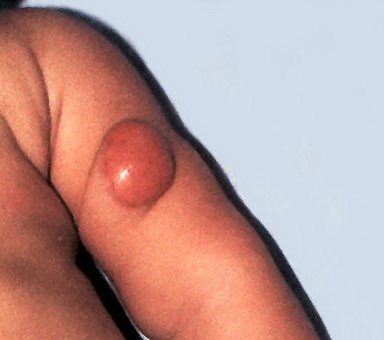
100) firm hemispheric lesions 2 to 5 mm in diameter. These lesions are irregularly scattered throughout the skin but are located mainly on the upper part of the body. PX is considered to be a form of JXG in which the lesions rapidly become xanthomatous. PX lesions range from 2 to 12 mm and show a generalized distribution with no tendency to merge into plaques. The nodular form is less frequent and occurs as one or a few lesions. Such nodules are generally round, 10 to 20 mm in diameter, and translucent, and might show telangiectases on their surface. The term giant juvenile xanthogranuloma is applied to lesions larger than 2 cm. Unusual clinical variants have been also reported. The mixed form is characterized by the simultaneous presence of both small and large nodules. The term juvenile xanthogranuloma en plaque defines a group of JXG lesions with a tendency to coalesce into a plaque as the only expression of the disease. In one study, 67 percent of 174 cases involved solitary papular lesions, and an additional 16 percent showed solitary nodular lesions. Multiple cutaneous lesions were noted in 7 percent of affected individuals, a solitary extra-cutaneous lesion in 5 percent, and multiple cutaneous and visceral-systemic lesions in an additional 5 percent.
RELATED PHYSICAL FINDINGS
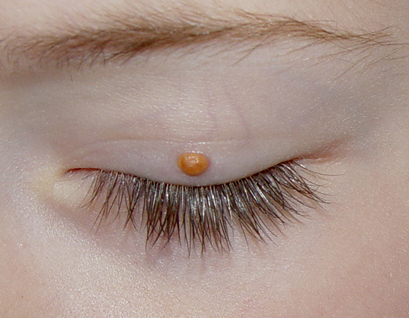
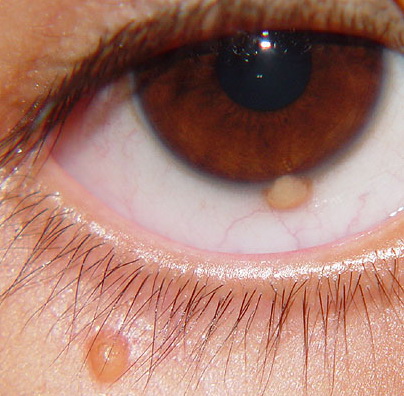
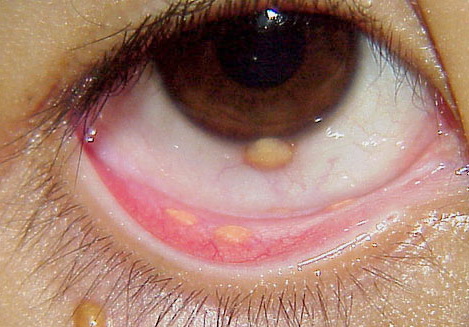
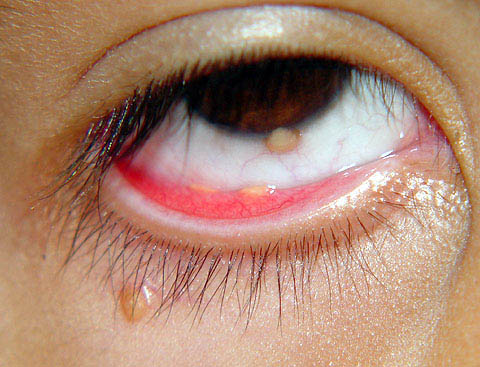
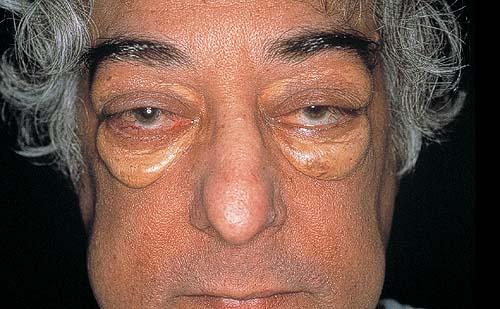
Ocular involvement is the most typical extra-cutaneous manifestation. It may precede or follow the cutaneous lesions. Ocular JXGs are usually unilateral and
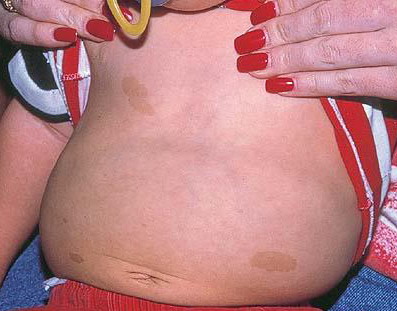
may lead to hemorrhage and glaucoma. An extremely rare extra-cutaneous manifestation of the papular variant is central nervous system (CNS) involvement. The nodular form of JXG may occasionally be related to systemic lesions of the lungs, bones, kidneys, pericardium, colon, ovaries, and testes. Juvenile chronic myelogenous leukemia has been observed in association with this variant of JXG. Café-au-lait macules and JXGs occur together in 20 percent of individuals with papular JXGs and can be considered an excellent marker of neurofibromatosis type 1 (NF-1) (see Chap. 142) during the first years of life, even in the absence at that age of other reliable markers of NF-1. Although the presence of JXGs in children has been said to be a marker of chronic myelogenous leukemia with and without concurrent NF-1, few children with café-au-lait macules and JXGs show evidence of leukemia.
Laboratory Tests
No abnormalities in laboratory test results are usually found in JXG except in the very rare cases in which juvenile chronic myeloid leukemia is associated with JXG.
Histopathologic and Ultramicroscopic Findings
Biopsy sections of JXG lesions show a feature that is common to many NLCHs, that is, a nonepidermotropic histiocytic infiltrate lacking Langerhans granules. Early lesions of JXG are characterized by a monomorphous, non-lipid-containing histiocytic infiltrate that occupies at least the upper half, and sometimes the entire thickness, of the dermis. Mature lesions
contain foam cells, foreign-body giant cells, and Touton giant cells, mainly distributed in the superficial dermis and on the border of the infiltrate. Lymphocytes, eosinophils, and neutrophils are variably associated. Older lesions may show fibrosis. In mature lesions, fat stains yield positive results. The majority of JXG lesional biopsy sections stain positive for CD68/Ki-M1P and factor XIIIa but negative for CD1a and S100 protein.
Under the electron microscope, the histiocytes that characterize the early stage of the disease exhibit pleomorphic nuclei, are rich in pseudopods, and contain many elongated and irregular dense bodies. Clusters of comma-shaped bodies can occasionally be observed. In older lesions, there is a predominance of foam cells, the cytoplasm of which is completely filled with lipid vacuoles, cholesterol clefts, and myeloid bodies. Touton giant cells are extremely large and sometimes contain more than 10 nuclei. Lipid material fills the periphery, whereas mitochondria and lysosomes predominate at the center.
The lesions of PX are composed almost entirely of foam cells and Touton giant cells. There is no evidence of a primitive histiocytic phase, and inflammatory cells are scarce or absent. Ultrastructurally, the cytoplasm is completely filled with lipid vacuoles without a limiting membrane, myeloid bodies, and lysosomal inclusions. Comma-shaped bodies are not found in these histiocytes.
Differential Diagnosis
The differential diagnosis of JXG is summarized in Box 149-1.
Complications
Eye involvement of a JXG lesion can cause a major visual problem leading to
severe secondary glaucoma. Lesions compatible with JXG have been described in many other organs as well, including lungs, spleen, liver, testes, pericardium, gut, kidney, and even the CNS, and in rare cares can lead to death. Patients may respond to multiagent chemotherapeutic regimens used to treat LCH.
Box 149-1 Differential Diagnosis of Juvenile Xanthogranuloma
|
PAPULAR
|
NODULAR
|
|
Most Likely
|
Most Likely
|
|
▪ Papular xanthoma
|
▪ Hashimoto-Pritzker disease
|
|
▪ Xanthoma disseminatum
|
▪ Nodular mastocytoma
|
|
▪ Tuberous xanthoma (hyperlipemic)
|
▪ Spitz nevus
|
|
▪ Sarcoidosis (lichenoid form)
|
|
|
▪ Papular mastocytosis
|
Consider
|
|
|
▪ Dermatofibroma
|
|
Consider
|
▪ Solitary cutaneous reticulohistiocytosis
|
|
▪ Benign cephalic histiocytosis
|
▪ Giant molluscum contagiosum
|
|
▪ Generalized eruptive histiocytosis
|
|
|
▪ Diffuse cutaneous reticulohistiocytosis
|
|
|
▪ Eruptive Spitz nevi
|
|
|
▪ Molluscum contagiosum
|
|
|
▪ Leukemia
|
|
Prognosis and Clinical Course
The cutaneous lesions of JXG tend to flatten with time. Each lesion evolves separately; thus, lesions at different stages of development may be seen in the same patient. Both cutaneous and visceral lesions disappear spontaneously, usually within 3 to 6 years. The patient's general health is not impaired, and physical and mental development is normal. Metabolic disturbances have not been identified. In the absence of associated conditions, prognosis is good.