X LINKED RECESSIVE ICHTHYOSIS
In the 1960s, X-linked recessive ichthyosis was distinguished clinically from other ichthyoses. Shortly thereafter, the syndrome of placental steroid sulfatase deficiency was described in pregnancies with failure to initiate labor in association with low maternal urinary estrogens. Because the majority of maternal urinary estrogens are derived from the fetal adrenal and are metabolized by the placenta, low levels can reflect fetal abnormalities or death. However, in this condition, low levels do not indicate severe fetal morbidity. The association between failure to initiate or progress labor and ichthyosis in the male offspring was not appreciated until later. Steroid sulfatase hydrolyzes sulfate esters, which include cholesterol sulfate and sulfated steroid hormones. Sulfated fetal adrenal hormones undergo desulfation to estrogens, which are excreted in maternal urine. The absence of steroid sulfatase enzyme in the fetal placenta leads to low maternal urinary estrogens and, in some pregnancies, to a failure of labor to initiate or to progress normally. In males with X-linked recessive ichthyosis, steroid sulfatase enzyme activity is decreased or absent in many tissues, including epidermis, stratum corneum, and leukocytes, and in cultured fibroblasts. In addition, cholesterol sulfate, an enzyme substrate, accumulates in serum and in scale. Carrier females have been found to have leukocyte steroid sulfatase levels intermediate between those observed in normal individuals and those in affected males.
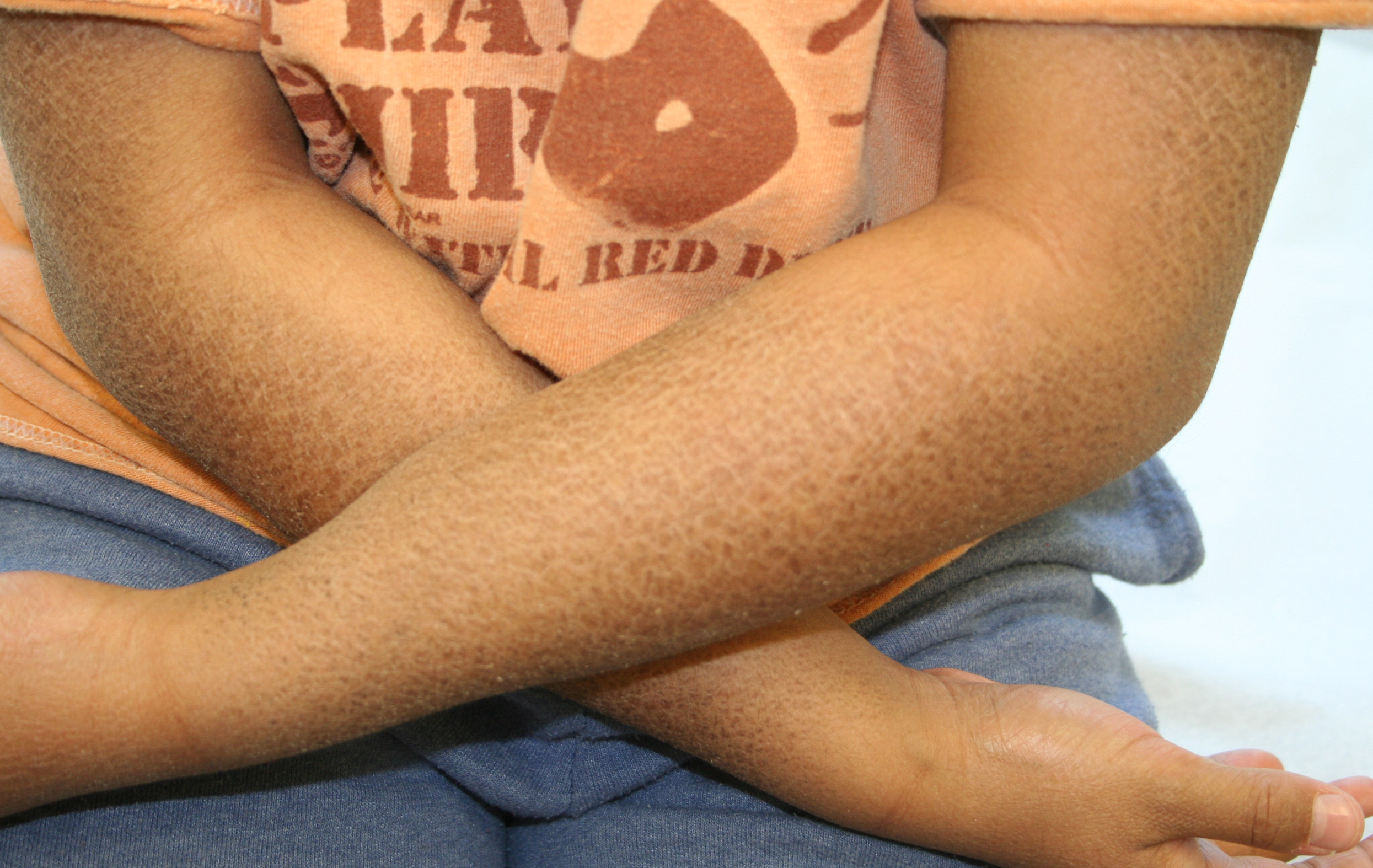
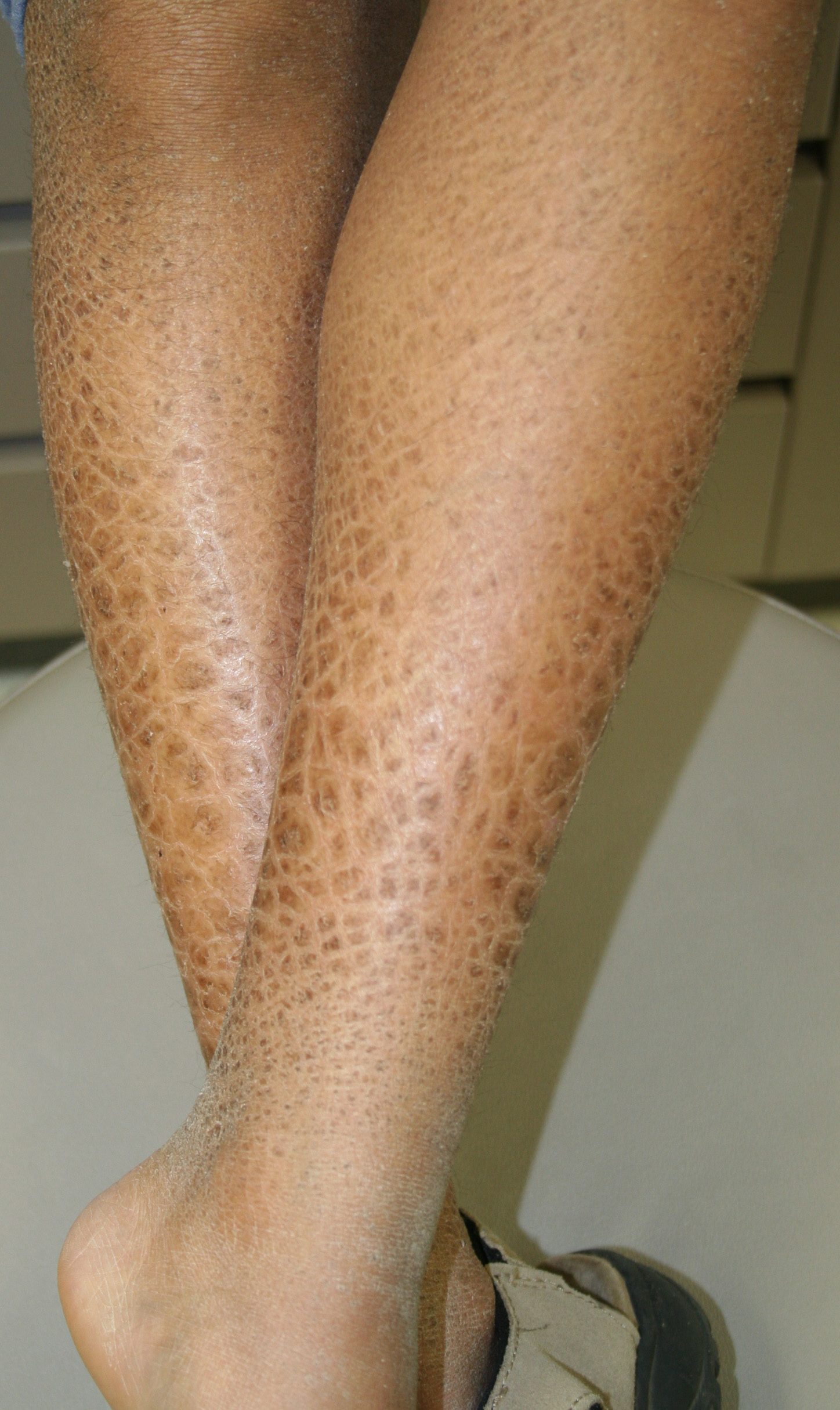
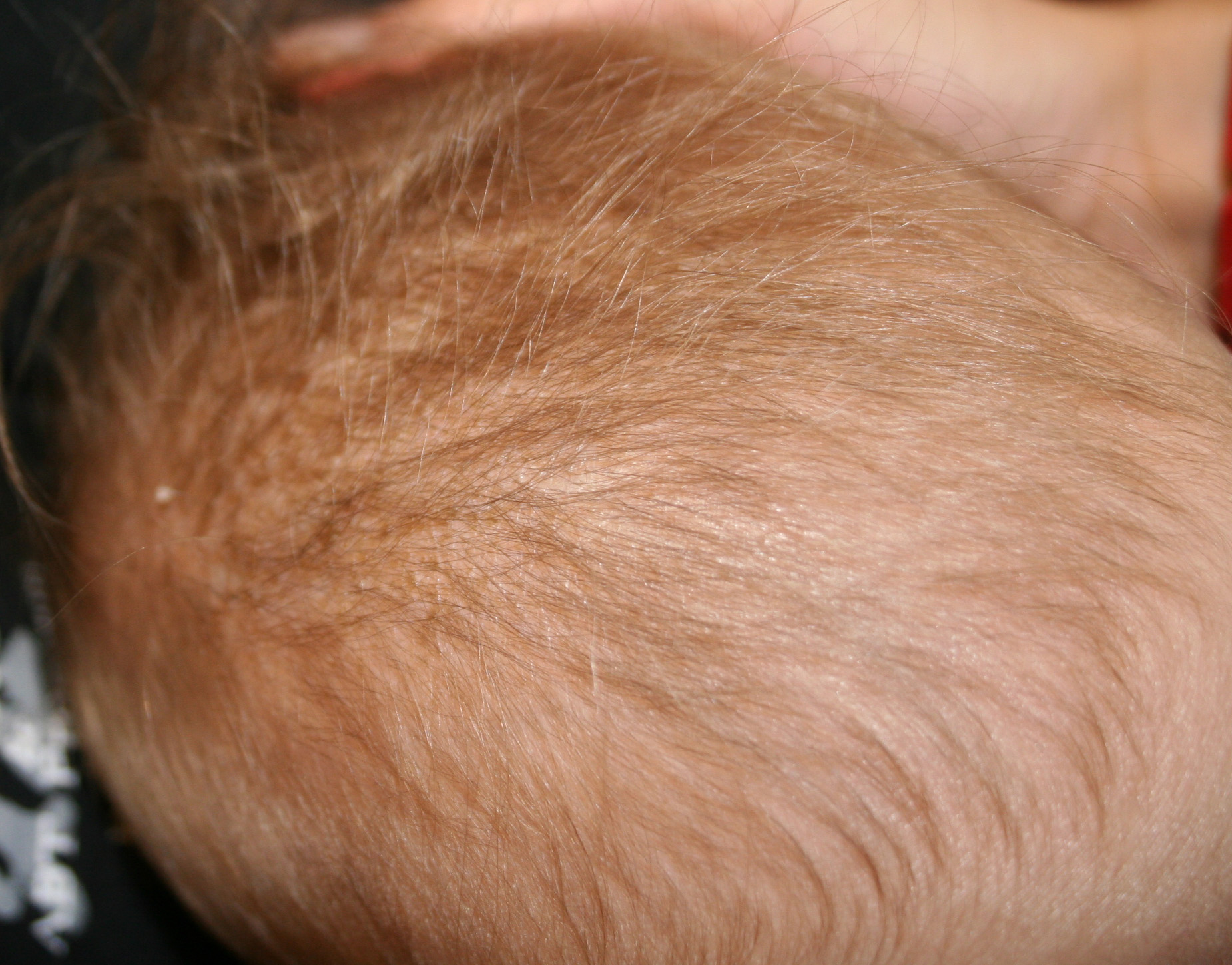
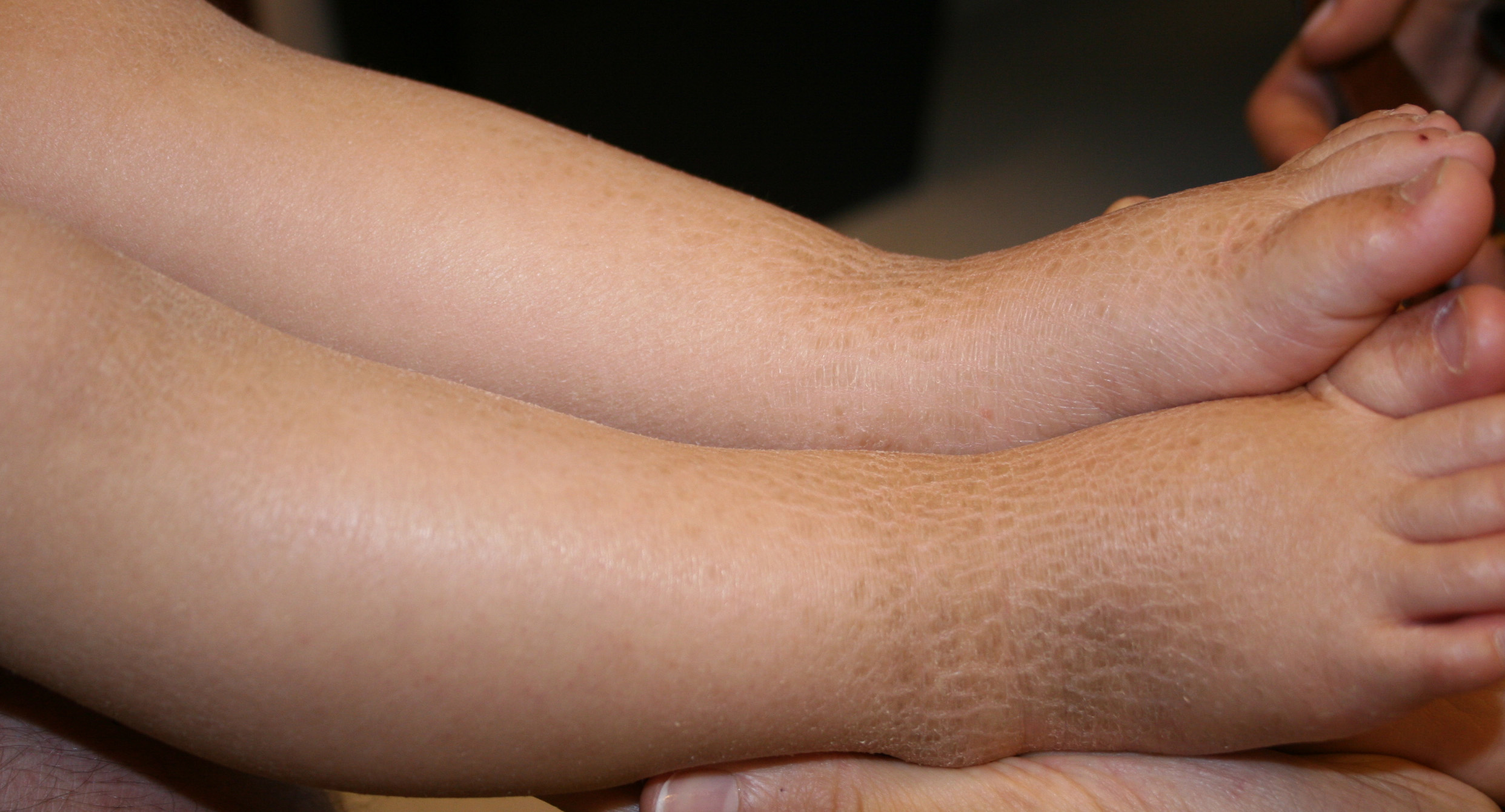
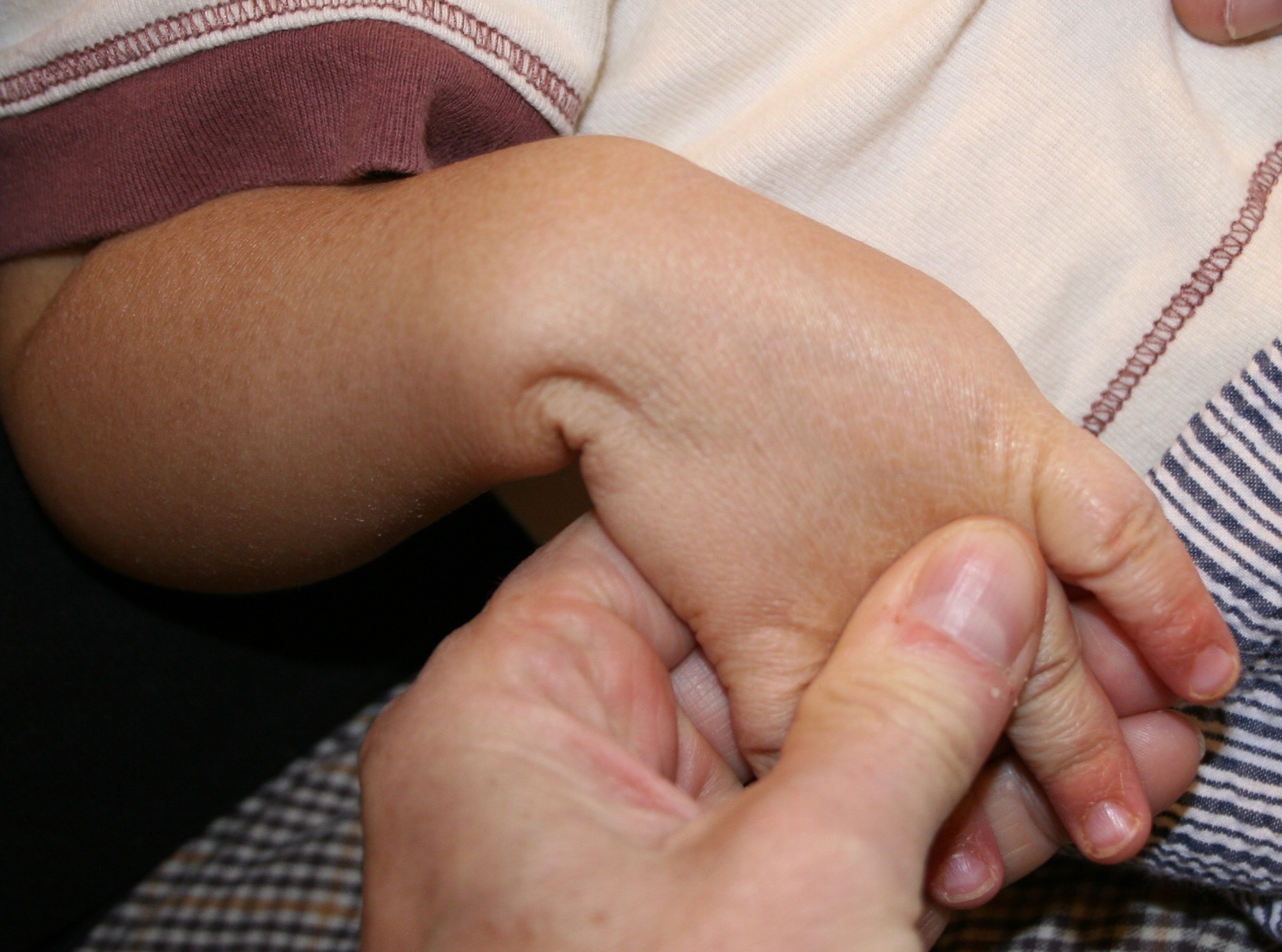
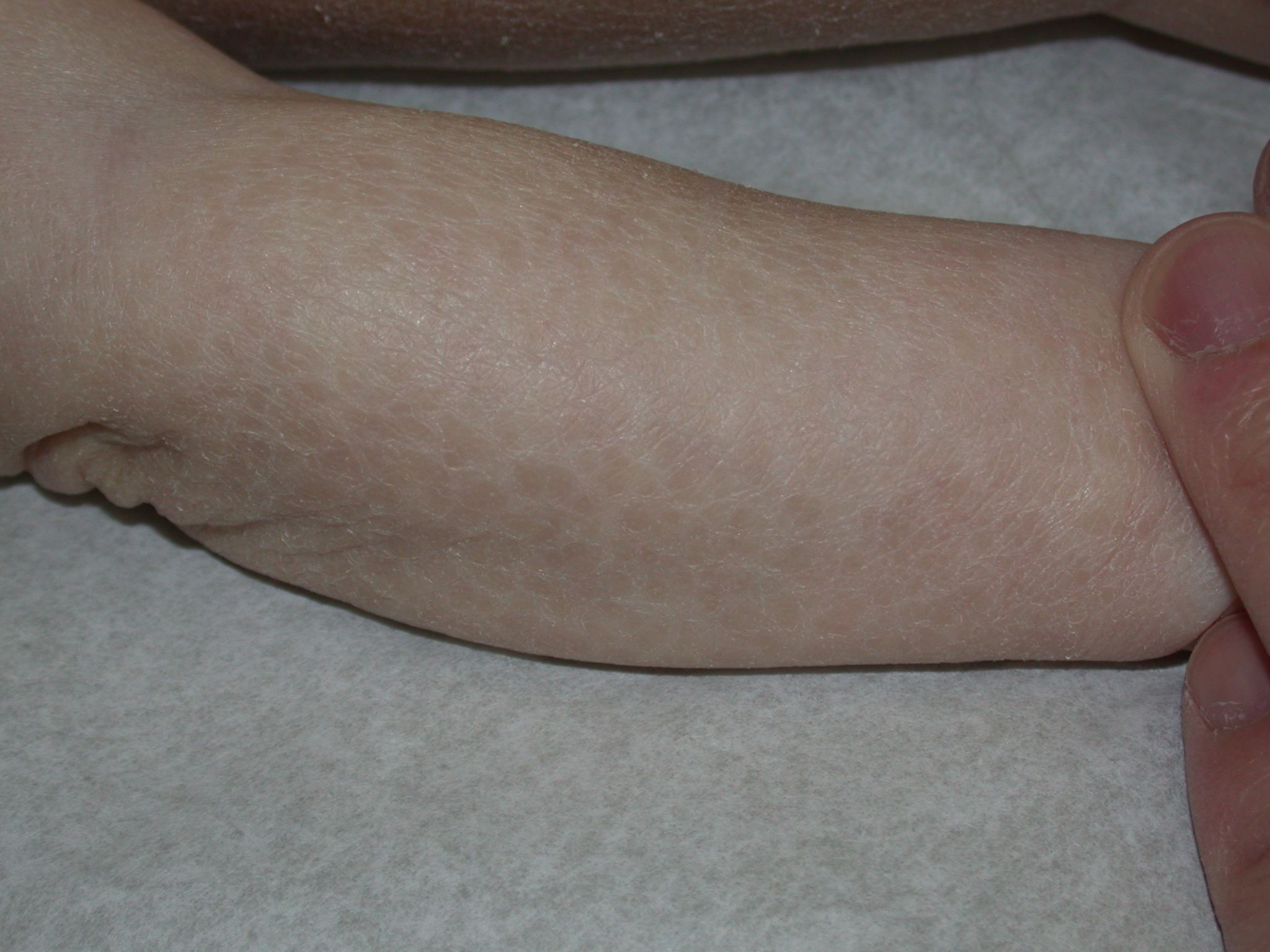
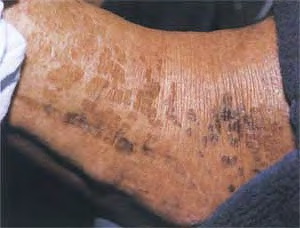
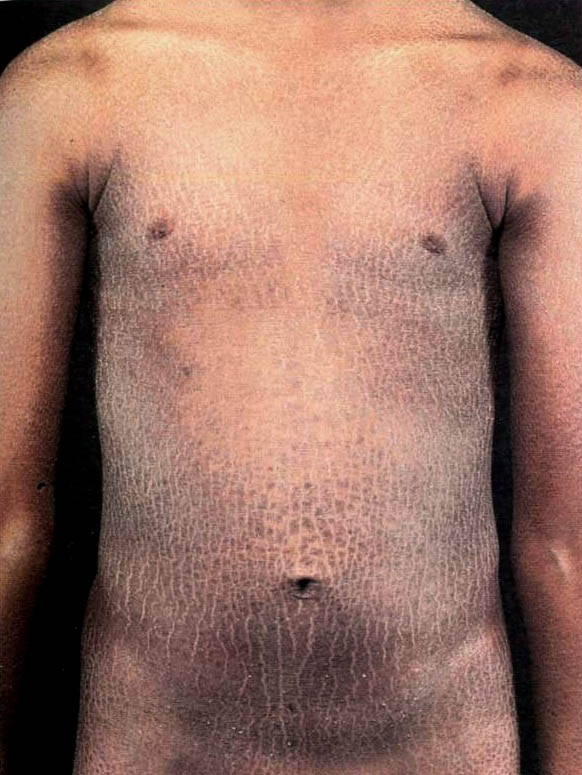
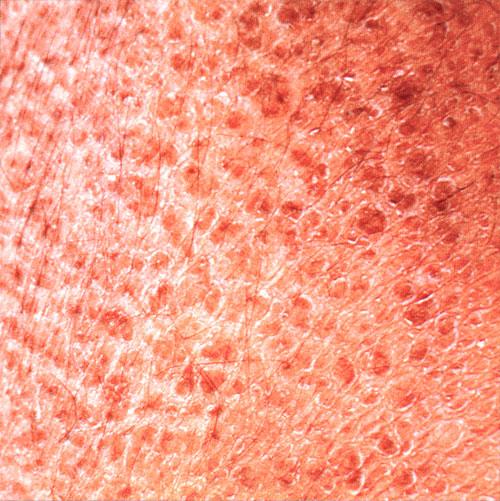
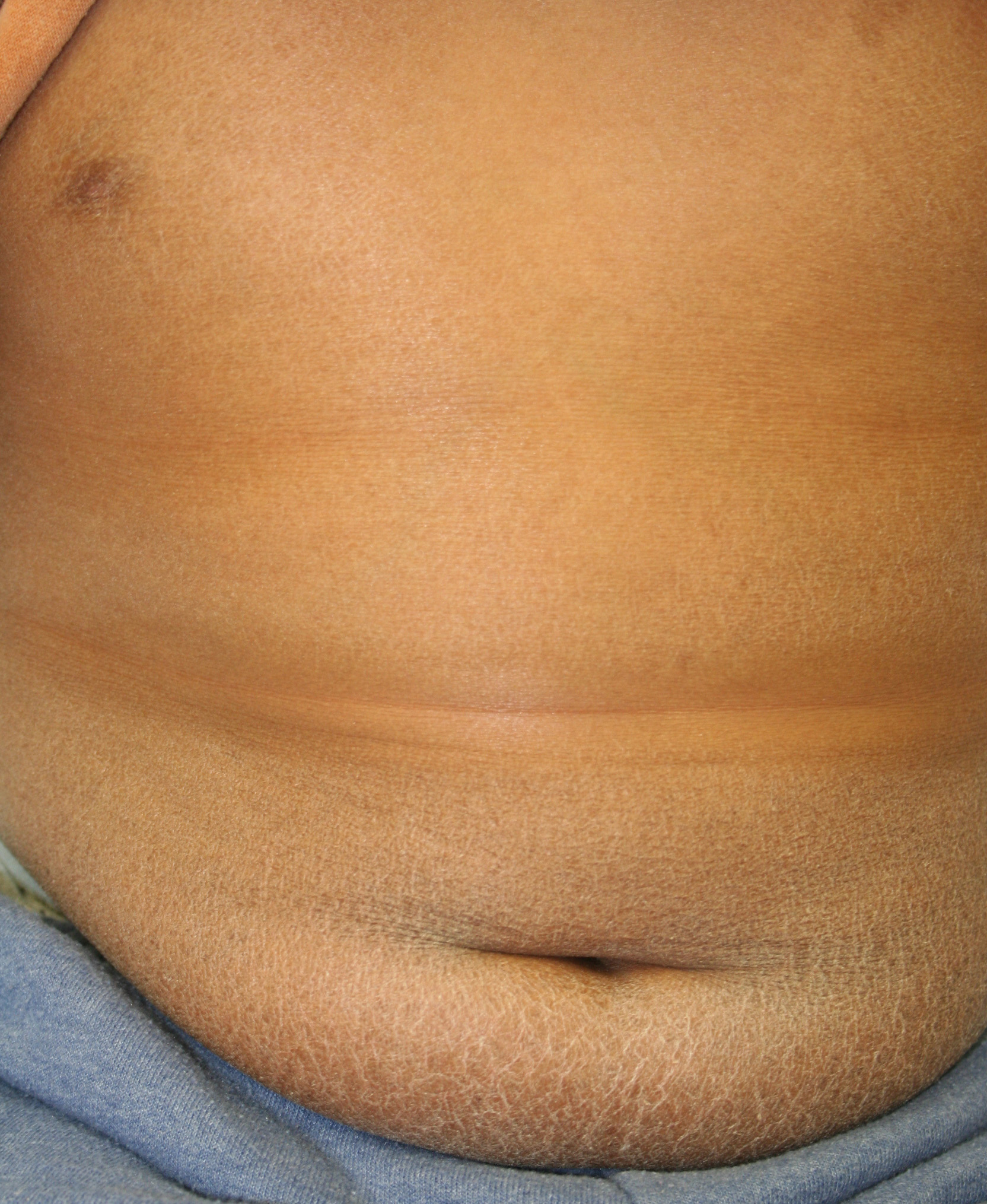
X-linked recessive ichthyosis occurs in approximately 1 in 2000 to 6000 males. Scaling may begin in the newborn period and is usually most prominent on the extensor surfaces, although there is significant involvement of the flexural areas. Although the extent and degree of scaling are variable, X-linked ichthyosis can usually be distinguished from ichthyosis vulgaris on clinical criteria. The latter tends to be associated with hyperlinear palms and soles, keratosis pilaris, and a family history of atopy. X-linked ichthyosis tends to have more severe involvement with larger scale, and comma-shaped, corneal opacities may be present in one-half of adult patients Corneal opacities do not affect vision and may be present in female carriers. Affected males have an increased risk of cryptorchidism and, independently, they are
at increased risk for the development of testicular cancer. Histopathologic examination shows compact orthohyperkeratosis, acanthosis, and papillomatosis. The granular layer is usually thickened .
Cholesterol sulfate levels are elevated in the serum, epidermis, and scale, and there is increased mobility of β-lipoproteins (low-density lipoproteins) on electrophoresis, a feature that can suggest the diagnosis. Steroid sulfatase is one of a group of arylsulfatases located on chromosome Xp22. More than 90 percent of the mutations in X-linked ichthyosis are deletions, explaining the failure to find immunologically detectable enzyme protein in some patients. Deletions can often be detected by fluorescence in situ hybridization, available in many clinical laboratories. Confirmation of the diagnosis can also be made by finding an elevation in serum cholesterol sulfate levels (normal is 80 to 200 µg/dL; X-linked ichthyosis is 2000 to 9000 µg/dL). Unfortunately, cholesterol sulfate determination in serum and scale may not be readily available for laboratory confirmation of the clinical diagnosis. In the rare case in which deletions are not detected and a clinical diagnosis is necessary, arylsulfatase C (steroid sulfatase) enzyme activity can be measured in cultured fibroblasts. Deletions that include adjacent sulfatases explain the overlap syndromes involving chondrodysplasia punctata and X-linked ichthyosis. The X-linked form of Kallmann syndrome, in which hypogonadotrophic hypogonadism and anosmia are found, often with renal abnormalities, obesity, synkinesis (mirror image movements of the extremities), cleft palate, and spastic paraplegia, can also be seen in association with X-linked recessive ichthyosis as part of a contiguous gene deletion syndrome. Because of this, and because of the association with testicular carcinoma, patients with X-linked recessive ichthyosis should be queried about anosmia and have periodic testicular examination.
In the epidermis, steroid sulfatase catalyzes the hydrolysis of cholesterol sulfate. The identification of steroid sulfatase deficiency in X-linked ichthyosis supports the importance of cholesterol sulfate hydrolysis in normal desquamation. Topical application of cholesterol sulfate in mice can induce a scaling disorder, further supporting the role of cholesterol sulfate hydrolysis in corneocyte desquamation.
A family was described with an ichthyosis inherited in an X-linked pattern but with normal steroid sulfatase activity and the absence of corneal opacities. This demonstrates heterogeneity within X-linked ichthyosis. Because of its frequent occurrence, steroid sulfatase deficiency accounts for most cases of X-linked ichthyosis, but a normal steroid sulfatase level in a male with ichthyosis does not rule out an X-linked pattern of inheritance.