WAARDENBURG
SYNDROME
WS, described by the Dutch physician Petrus Waardenburg in 1951, is the prototypic congenital disorder of pigmentation. Although it was originally described as a syndrome combining pigmentary defects of the hair (poliosis, or white forelock) and iris, congenital deafness, and developmental craniofacial abnormalities, it was subsequently realized that additional phenotypic manifestations could be part of the same syndrome. Four types of WS, WS1 through WS4, have been described. The discovery of molecular mutations accounting for the different types of WS has helped to explain its wide variety of manifestations as well as to illuminate the importance of specific genes for the development of different tissues and organs. Although practically all cases of WS1 and WS3 show mutated PAX3, WS4 individuals either have homozygous mutations in EDN3 (the endothelin-3 gene) or EDNRB (the endothelin B receptor gene) or heterozygous mutations in SOX10. On the other hand, WS2 appears to arise more heterogeneously, because a mutation in MITF has been shown in only a small fraction of WS2 patients. In addition to MITF, SLUG/SNAI2, another transcription factor gene, was found to be mutated in two unrelated individuals with WS2.
Waardenburg Syndrome
Type1.
Individuals with WS1 are usually heterozygous for mutations in PAX3; hence, WS1 is autosomal dominant in inheritance. Although many different mutations in PAX3 have been associated with WS1, these mutations are thought either to be functionally null alleles or to abrogate the interactions of PAX3 with DNA.
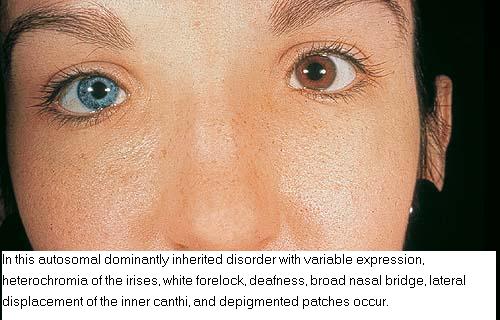
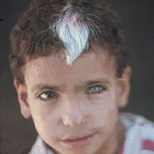
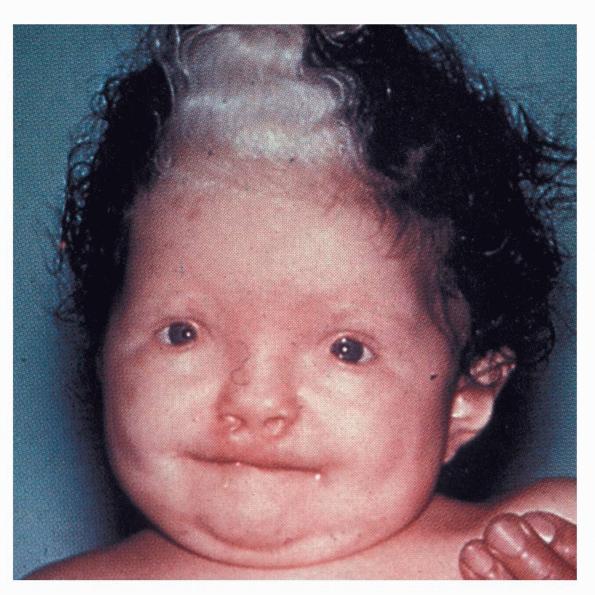
Individuals with WS1 have pigmentation abnormalities associated with craniofacial abnormalities . Dystopia canthorum, which is lateral displacement of the medial canthi of the eyes, is the hallmark craniofacial defect found in virtually all cases of WS1. A broadening of the nasal root, the presence of hypoplastic alae nasi, and synophrys are other craniofacial abnormalities associated with WS1. Poliosis, such as the presence of a white forelock, is the most common pigmentation abnormality associated with WS1. However, a wide variety of other pigmentary abnormalities can also be associated with WS1, albeit less frequently. These include depigmented white spots on the skin and pigmentary abnormalities of the iris. Iris pigmentary abnormalities may include complete heterochromia irides (differently colored irises), partial heterochromia irides (variations of color within an iris), or hypoplastic blue irides. Premature graying may also be associated with WS1. Congenital deafness is present in 57 percent of cases.
The importance of PAX3 for the expression of MITF, with consequent effects on melanocyte survival during development, is likely to account for the pigmentary defects of WS1. A role for PAX3 in governing the development of neural crest derivatives that contribute to bony and cartilaginous structures of the face, particularly those contributing to the formation of the frontal bone, explain the craniofacial anomalies observed in WS1. Sensorineural deafness, observed with incomplete penetrance in WS1, results from the variable failure of melanoblasts to migrate to or to survive in the stria vascularis in the lateral wall of the cochlea.
Waardenburg Syndrome
Type2.
Linkage analysis of families with WS2 identified the MITF locus as a candidate locus for the disease gene. At least nine different mutations have been found in the coding region of the MITF gene in WS2 families. However, MITF mutations account for only a minor portion (15 percent) of WS2 cases. A mutation in the transcription factor gene SLUG/SNA12 has also been associated with WS2, but mutations in other, as yet undefined genes are likely to be implicated in the future. WS2 is inherited in an autosomal dominant pattern. Because most of the known MITF mutations in WS2 compromise the HLHZip region, thereby interfering with dimerization of mutant MITF with wild-type MITF, the pigmentary developmental pathology in most cases of WS2 probably occurs through haploinsufficiency (reduced gene dosage, expression, or protein activity) rather than through dominant-negative effects. Tietz syndrome, described later, is likely to result from dominant-negative effects of mutant MITF.
Although all types of WS have skin, hair, and iris pigmentation anomalies and the possibility of hearing loss, WS2 is notable for featuring only these auditorypigmentary symptoms. Diagnostic criteria for WS2 have previously been defined. Individuals fulfilling two of the following four criteria, in the absence of dystopia canthorum, limb deformity, or Hirschsprung disease, should be counted as affected:
· Congenital sensorineural hearing loss
· Pigmentary disturbance of iris
o Complete heterochromia irides (two eyes of different color)
o Partial or segmental heterochromia (segments of blue or brown pigmentation in one or both eyes)
o Hypoplastic blue eyes (characteristic brilliant blue in both eyes)
· Pigmentary disturbance of the hair
o White forelock from birth or in teens
o Premature graying before age 30 years
· A first- or second-degree relative with two or more of criteria 1 to 3
A survey of 124 cases of WS2 and 270 cases of WS1 revealed differences of phenotypic penetrance between WS2 and WS1: congenital sensorineural hearing loss occurred in 77 percent and 57 percent, respectively; heterochromia irides in 48 percent and 27 percent, respectively; hypoplastic blue eye in 9 percent and 17 percent, respectively; white forelock in 9 percent and 17 percent, respectively; early graying in 23 percent and 26 percent, respectively; and white skin patches in 6 percent and 31 percent, respectively. The higher incidence of hearing loss in WS2 may be due to the difficulty of diagnosing WS2 in individuals without hearing loss. The hearing loss is congenital, sensorineural, and nonprogressive, showing marked variation between and within families. Among a series of 81 WS2 cases studied by these investigators, 84 percent of patients reported bilateral hearing loss; 40 percent of patients noted profound loss, whether unilateral or bilateral.
Waardenburg Syndrome
Type3.
WS3 , also known as Klein-Waardenburg syndrome, is regarded as a variant of WS1. Most affected persons are heterozygous for a mutation in PAX3, although a few severely affected homozygotes have been described. No specific mutations in PAX3, with the possible exception of a missense mutation at Asp47, have been correlated with the WS3 phenotype rather than the WS1 phenotype. In addition to the features of WS1, WS3 patients have musculoskeletal abnormalities, manifested as limb contractures and hypoplasia of the limb musculature. The WS3 phenotype is consistent with the previously described role of PAX3 in the activation of transcription factors that govern muscle and limb development, distinct from its role in regulating the development of neural crest derivatives.
Waardenburg Syndrome Type 4.
WS4 , also known as Shah-Waardenburg syndrome, is caused by heterozygous mutations in the transcription factor gene SOX10 or by homozygous mutations in the gene encoding the peptide ligand endothelin-3, EDN3, or its receptor, EDNRB.137 In addition to governing aspects of melanocyte development, these genes are important determinants of the development of the distal aspect of enteric nervous system cells, also neural crest derived, that innervate the distal part of the colon, which explains the associated Hirschsprung disease (megacolon). WS4 is the combination of the WS1 phenotype with Hirschsprung disease, or congenital aganglionosis of the colon