| Pct = البورفيريا الجلدية الاجلة |
|
|
Porphyria Cutanea Tarda
EPIDEMIOLOGY PCT occurs throughout the world and is the most common of all the porphyrias. The prevalence is estimated at 1 in 10,000. The disease most often begins in middle-aged individuals but can develop earlier. Before the widespread use of oral contraceptives, PCT developed predominantly in males. Others, however, have emphasized that the sex incidence is now approximately equal. The rising incidence of PCT in females is probably due to the widespread ingestion of estrogens in oral contraceptives or in hormone supplements. It should be noted that males treated with estrogens, for example as adjunctive therapy for carcinoma of the prostate, have also developed PCT. ETIOLOGY AND PATHOGENESIS PCT is due to either an inherited or an acquired deficiency of UROGEN decarboxylase, the fifth enzyme in the heme pathway , which has a molecular mass of approximately 42 kd.51 UROGEN I and III each contain eight carboxyl groups as side chains, four of which are acetate (-CH2COOH) and four of which are propionate (-CH2-CH2-COOH) moieties . The soluble cytosolic enzyme UROGEN decarboxylase catalyzes the sequential oxidative decarboxylation of the four carboxyl groups of the acetate side chains to methyl groups to form COPROGEN . Decarboxylation first occurs on ring D, after which the enzyme turns around to decarboxylate rings A, B, and C in a clockwise fashion. This converts the original 8-carboxyl porphyrinogen (UROGEN I or III) first to 7-carboxyl, then to 6-carboxyl and to 5-carboxyl porphyrinogen. The 5-carboxyl porphyrinogen can then undergo decarboxylation of its last acetyl group to form the 4-carboxyl porphyrinogen that is known as COPROGEN I or III . These intermediates are also referred to as hepta-, hexa-, penta-, and tetracarboxylate porphyrinogens. COPROGEN I cannot be further metabolized to heme.
Human UROGEN decarboxylase is encoded by a single-copy gene of the same name, UROGEN decarboxylase, that is located on chromosome 1p34, spreads over a genomic distance of 3 kilobases (kb), and contains 10 exons.51 The human UROGEN decarboxylase cDNA has been isolated, and the deduced sequence was found to be equivalent to 367 amino acids, consistent with the molecular mass and the amino acid composition of the purified protein. Most classifications of PCT separate the disorder into at least two broad categories, both associated with decreased UROGEN decarboxylase activity: (1) acquired PCT, also referred to as sporadic or type I PCT; and (2) hereditary PCT, also referred to as familial or type II PCT. In acquired (type I) PCT, the enzyme is deficient only in the liver, which could be explained either by the presence of a different gene defect restricted to the liver or by exposure to chemicals that selectively inhibit the hepatic but not the RBC enzyme. Some of these substances (e.g., alcohol and estrogen) may provoke PCT only in selected individuals and others (e.g., hexachlorobenzene or HCB) in practically all exposed individuals. Hereditary (type II) PCT is an autosomal dominant disorder, and the residual UROGEN decarboxylase activity is decreased approximately 50 percent in all tissues, including RBCs and cultured skin fibroblasts.53-56 Decreased enzyme concentration appears to follow a bimodal distribution, which suggests two overlapping groups of patients: A large group (more than 80 percent) with normal UROGEN decarboxylase concentration and a small group (fewer than 20 percent) in whom the amount of detectable enzyme is about half the normal level. Some patients with type II PCT were found to have UROGEN decarboxylase activity at the lower end of the normal range. The clinical penetrance of type II PCT is relatively low (approximately 20 percent), so that the majority of individuals with the inherited enzyme defect do not manifest the disease. This suggests that additional genetic or non-genetic factors are needed for disease expression. To date, more than 100 different mutations in the UROGEN decarboxylase gene have been identified that either decrease the stability of the enzyme or produce defective pre-mRNA splicing. It is important to emphasize that not every patient with a positive family history of PCT will have type II disease. In support of this notion, several patients have been described who have one or more relatives with PCT but have normal RBC UROGEN decarboxylase activity. This latter category of PCT has been designated as type III by some investigators.54 Either these patients could have inherited some form of UROGEN decarboxylase that is immunochemically indistinguishable from the normal enzyme but that is uniquely susceptible to inhibition in the liver, or they could have a second inherited enzyme deficiency unrelated to UROGEN decarboxylase. These possibilities require further investigation. It is likely that some patients who were initially reported as having hereditary PCT actually had VP, another dominantly inherited porphyria. PCT and VP may occur in different members of the same family in the so-called dual porphyrias. Another form of dual porphyria in which PCT and AIP co-exist has also been described. Numerous agents and conditions are known to contribute to the development of PCT (type I, acquired, or sporadic), including alcohol; estrogens; iron; viral infections [hepatitis C and human immunodeficiency virus (HIV) infection]; polychlorinated hydrocarbons, particularly HCB and 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD); and hemodialysis in patients with renal failure . Each of these precipitating factors is discussed briefly in the following sections. Alcohol. Alcohol ingestion has long been recognized to exacerbate PCT. Ethanol has been shown to induce hepatic ALA synthase in patients with PCT.65 Erythrocyte UROGEN decarboxylase activity is diminished in healthy individuals after acute ethanol ingestion and in those with chronic alcoholism.66 Ethanol can also inhibit the activity of other enzymes in the heme pathway, including ferrochelatase and
Estrogens. The widespread use of estrogens as contraceptive agents or as hormone supplements for post-menopausal hormone replacement therapy in females and as adjunctive hormonal therapy in males with prostatic carcinoma has been associated with the development of PCT.50 The mechanism of the estrogen effect on the expression of PCT has not been elucidated. Although diethylstilbestrol, an estrogen, induces hepatic ALA synthase,69 this would not explain the distinctive porphyrin excretion pattern found in PCT. The vast majority of patients receiving estrogens do not manifest the biochemical abnormalities associated with PCT. Hexachlorobenzene. The fungicide HCB caused an “epidemic” of a PCT-like syndrome in southeastern Turkey in the 1950s.70 It was added as a preservative to wheat intended for planting, but because of a famine, several thousand individuals of diverse ethnic origin, mostly children, ingested the seed wheat and subsequently developed typical PCT. Over 4000 cases of this syndrome were reported between 1956 and 1961. The porphyrin excretion pattern and the cutaneous findings in these patients were quite similar to those seen in PCT evoked by ethanol or estrogens. The outbreak of PCT in Turkey caused by ingestion of HCB indicated that the disease could occur in non-genetically predisposed individuals. Twenty-five years later, the most common clinical findings in these HCBpoisoned individuals were those of chronic porphyria, including sclerodermoid scarring (84 percent), hyperpigmentation (78 percent), hirsutism (49 percent), thyromegaly, and increased skin fragility (38 percent). A painless arthritis was seen in two-thirds of affected individuals, and a variety of neurologic signs and symptoms occurred in the majority. Stool and urine porphyrin levels remained elevated in many patients. Studies have shown that the long-term administration of HCB to experimental animals produces excessive porphyrin accumulation in the liver in a pattern quite similar to that seen in PCT in humans.72 These data are consistent with the hypothesis that chlorinated hydrocarbons such as HCB, or their metabolites, inhibit hepatic UROGEN decarboxylase, which leads to excessive hepatic storage of URO and other acetate-substituted porphyrins. Experimental studies have also shown that HCB can inactivate UROGEN decarboxylase and thereby abolish catalytic activity without changing the amount of immunoreactive enzyme protein. Chemical porphyria similar to PCT is caused by other chlorinated hydrocarbons such as the polychlorinated biphenyls and TCDD, a byproduct in the synthesis of the herbicide 2,4,5-trichlorophenoxyacetic acid. Additional studies on the porphyrinogenic effects of chlorinated hydrocarbons suggest that metabolic activation of the compounds mediated by cytochrome P 1A2 and involving iron-generated ROS is associated with an attack on the catalytic site of UROGEN decarboxylase. Tetrachlorodibenzo-p-Dioxin. TCDD is a toxic environmental pollutant. Among its numerous effects are chloracne, liver damage, and hepatic porphyria in experimental animals and perhaps also in humans.78 It has been shown that the hepatic porphyrinogenic effect of TCDD can be abolished in mice by first depleting them of iron.79 Furthermore, it is known that highly inbred mouse strains vary in their susceptibility to induction of hepatic porphyria by TCDD, which indicates that the porphyrogenic effect of this hydrocarbon is modulated by as yet undefined genetic factors. Iron. Serum iron and ferritin concentrations are elevated or in the upper range of normal in individuals with PCT, which confirms the important role of iron in the pathogenesis of the disease. Hepatic iron overload accompanies clinical PCT in practically all cases, and elevation of plasma iron level is found in one-third to one-half of patients. In PCT, the quantity of iron that can be mobilized by phlebotomy indicates that total iron stores are approximately twice the normal level. Ferrokinetic studies in patients with PCT are said to yield normal results. The long remissions that follow repeated phlebotomy and the apparent ineffectiveness of this treatment if supplemental iron is administered concomitantly suggest that iron plays a role in the excessive hepatic porphyrin production in PCT. PCT is particularly common when alcoholism and iron overload occur together. The role of iron in the pathogenesis of PCT is complex, and several hypotheses have been proposed to explain it. Iron may directly inhibit UROGEN decarboxylase. However, studies using purified UROGEN decarboxylase prepared from human erythrocytes show that the purified enzyme is not inhibited by Fe2+ or Fe3+ ions. Chronic iron overload can produce peroxidative damage to lipidrich mitochondrial and microsomal membranes in the liver of experimental animals, but the relationship of this toxic effect to changes in hepatic heme synthesis has not been clearly defined.84 An increased frequency of the hemochromatosis C282Y mutation in a gene encoding for a human leukocyte antigen class I-like protein, HFE, has been found in British patients with sporadic PCT.85,86 This mutation is responsible for much of the iron overload in populations of European descent. A second mutation in the HFE gene, H63D, may also be associated with PCT in some populations. PCT is rare in heterozygotes for each of these mutations, but 20 percent of British patients with PCT are C282Y homozygotes. C282Y homozygosity is an important susceptibility factor for both type I and type II PCT. Iron may have a permissive effect on the inhibition of UROGEN decarboxylase by halogenated hydrocarbons, and it can also enhance the induction response of hepatic ALA synthase to drugs. Although such an iron-augmented increase in ALA synthase activity could lead to enhanced porphyrinogenesis, this alone would not explain the porphyrin excretion pattern seen in PCT. Kushner, Lee, and Nacht have shown that the addition of ferrous iron to liver in vitro causes a marked increase in porphyrin synthesis and inhibits UROGEN III synthase activity, which provides an explanation for the URO isomer I excess that is characteristic of PCT. More recently, a mouse model of type II PCT was developed in which one allele of UROGEN decarboxylase is disrupted, and these animals were then bred to mice homozygous for HFE disruption. These animals developed a PCT-like phenotype. Viral Infections. There is an association between PCT and hepatitis C virus (HCV) infections and combined HCV and HIV infections. The role of these viruses in the pathogenesis of PCT
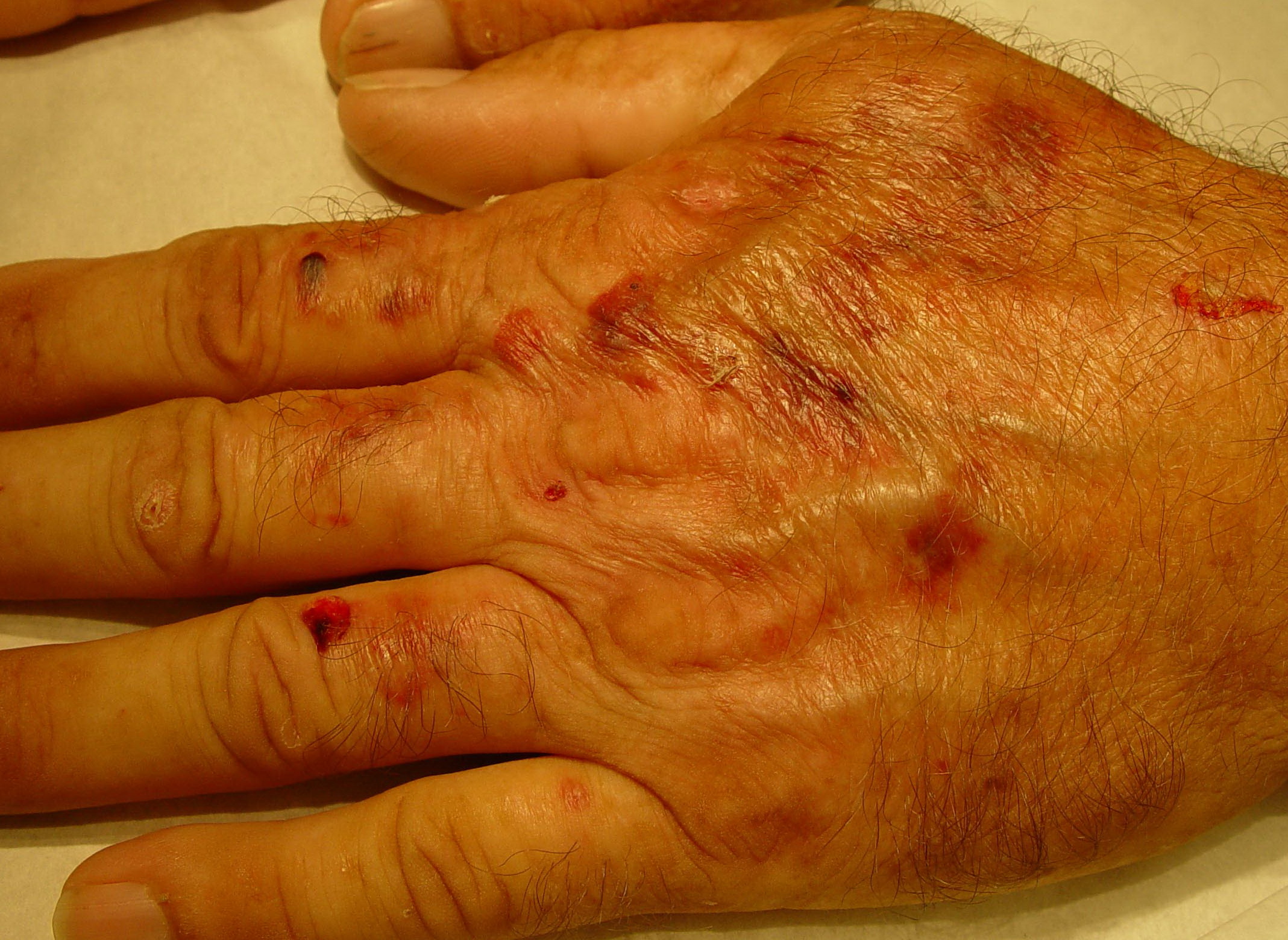
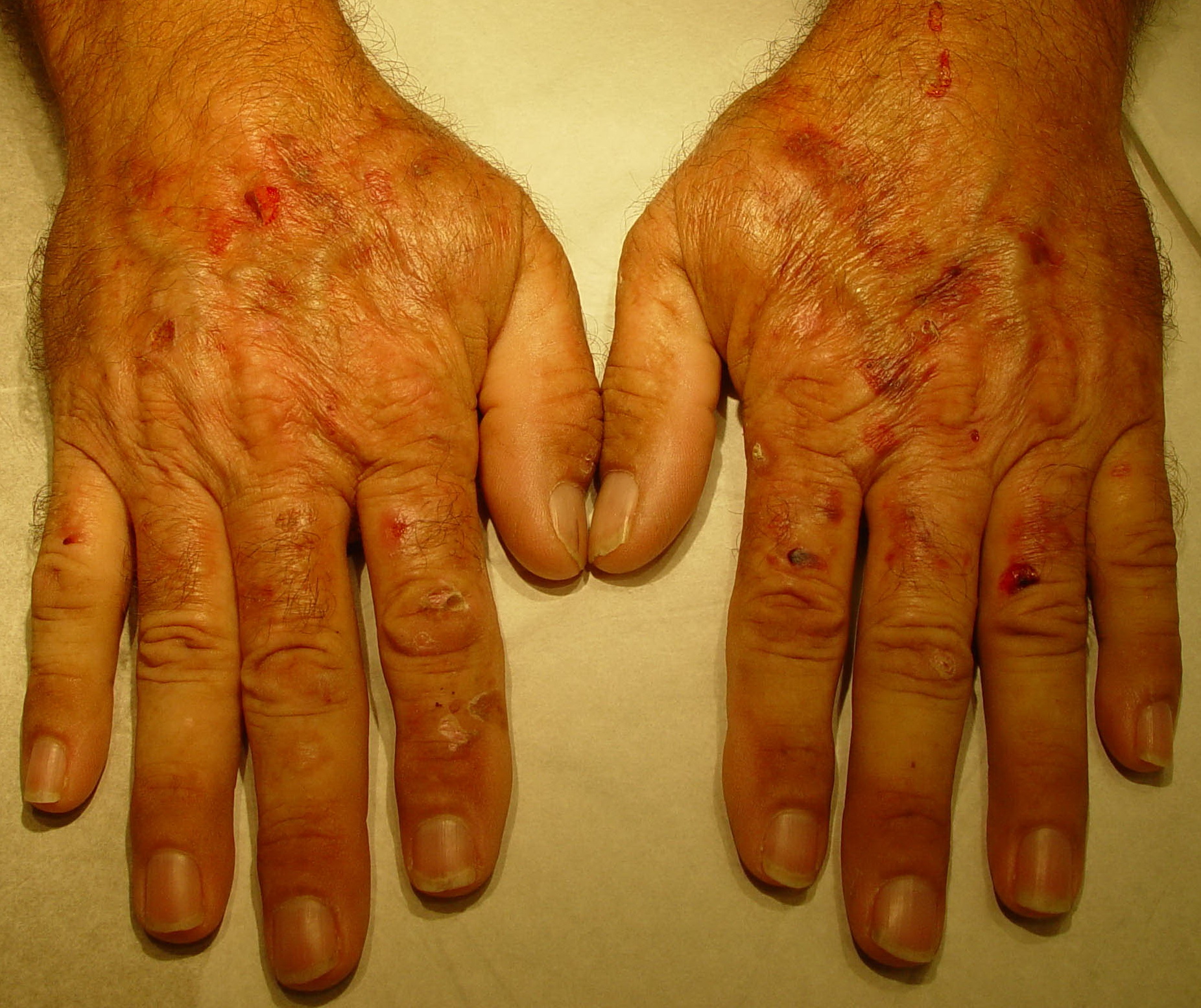
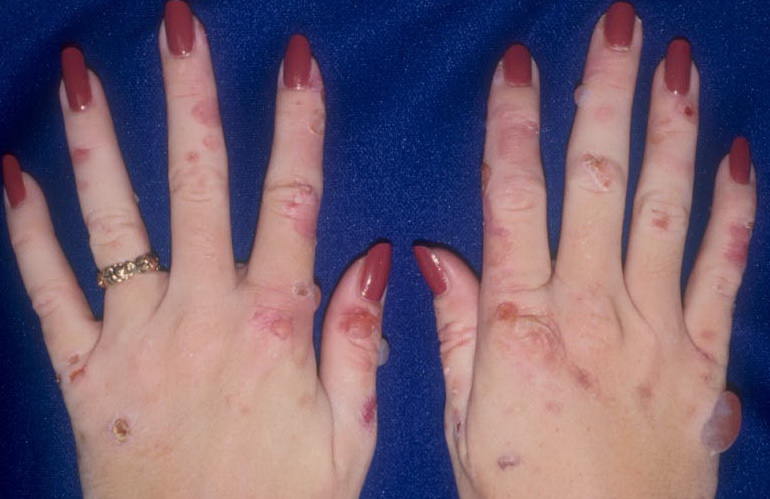
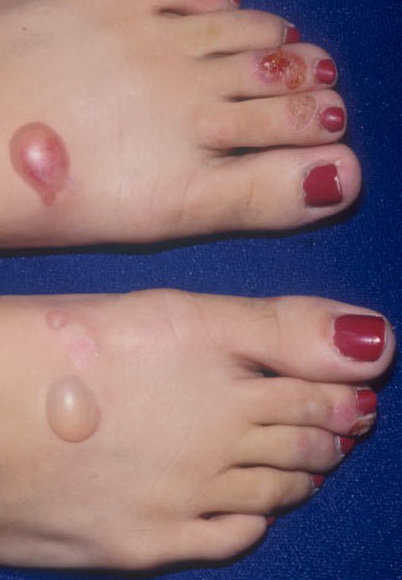
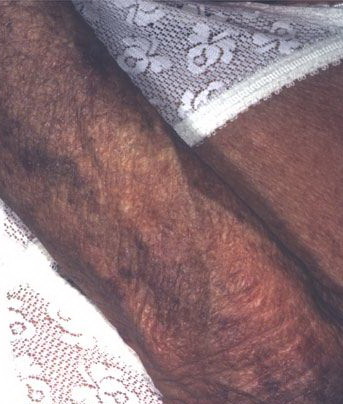
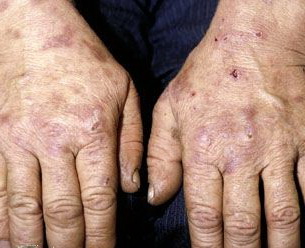
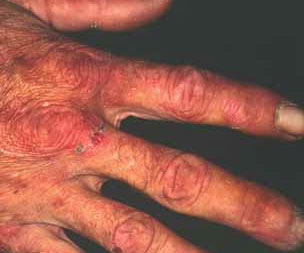
From these known effects of alcohol, estrogens, chlorinated hydrocarbons, iron, and viral infections on the heme pathway, it is clear that each of these could contribute to the excessive hepatic porphyrinogenesis characteristic of PCT. There is growing evidence that hepatic siderosis is the critical pathologic endpoint in PCT and that the other agents somehow intensify the ability of iron to attack the catalytic site of UROGEN decarboxylase. The clinical expression of PCT is therefore dependent on the interaction of a number of factors, both genetic and environmental. However, it is important to note that the ingestion of drugs that have been associated with the induction of acute neurovisceral attacks in the acute porphyrias is not known to exacerbate similar neurologic crises in PCT. CLINICAL MANIFESTATIONS Vesicles and bullae followed by erosions and crusting occur predominantly in areas subject to repeated trauma . There is increased skin fragility, usually on the dorsa of the hands. The traumatized skin becomes crusted, and as the lesions resolve, areas of scarring may ensue. Numerous small milia can develop, particularly on the fingers and hands. These are pearly white to yellow in each of the hepatic porphyrias associated with cutaneous photosensitivity (PCT, VP, and HCP) . Although the cutaneous lesions are seen primarily on the light-exposed areas, patients are often unaware that sunlight plays a role in producing their lesions, because the acute painful and burning photosensitivity so characteristic of the erythropoietic porphyrias is rare in PCT. However, most patients do recognize that their skin condition worsens in the spring and summer and seems to improve in the fall and winter. Porphyrin excretion in PCT appears to increase in the summer months and decrease in the winter months. Other skin changes seen in PCT include hyperpigmentation and hypopigmentation that may be mottled, resembling chloasma. There may be an associated purplish red (“heliotrope”) suffusion of the central part of the face, particularly involving the periorbital areas, which may bear a striking resemblance to the plethora seen in polycythemia rubra vera . This is not seen in the porphyrias of bone marrow origin. Hypertrichosis (non-virilizing) is a useful diagnostic sign that often brings the female patient to the dermatologist . Facial hypertrichosis develops gradually and is more apparent in females. The hair may vary in texture from fine to coarse and in color from light to dark. These hairs are particularly prominent along the temples and the cheeks but may occasionally involve the trunk and extremities in severe cases. Such hair may continue to grow, darken, and thicken, particularly on the cheeks, the forehead between the eyes, and at the hairline of the scalp. Males may complain that shaving is more difficult and that the growth pattern of their beard has changed. Hypertrichosis may be the presenting symptom in women, and a particularly severe form of hypertrichosis may occur in younger children with PCT and HEP. In the reports of HCB poisoning in Turkey, some of the children were described as “monkey-like” because of marked hypertrichosis. The mechanism of this phenomenon is unknown; androgen levels are reported to be normal. It is possible that surface receptors or growth factors for hair bulb keratinocytes are activated by the dual action of light and porphyrins. The hypertrichosis of PCT usually improves slowly after depletion of excessive hepatic iron stores. Sclerodermoid plaques can occur in PCT and in HEP and typically develop on both light-exposed and light-protected body areas .
PCT-like syndromes are occasionally seen in association with hepatic tumors and lupus erythematosus. Sub-epidermal bullous dermatoses mimicking PCT clinically and histologically have been described (see Pseudoporphyria). A number of cases of true PCT have occurred in patients with renal failure undergoing hemodialysis.97 Confirmation of the diagnosis rests on detection of markedly elevated plasma levels of porphyrins (usually 5- to 100-fold) and increased levels of isocoproporphyrin (ISOCOPRO) in the feces. LABORATORY FINDINGS Patients with PCT excrete increased amounts of porphyrins in the urine, which rarely may exhibit characteristic pink-red fluorescence when examined with a Wood's lamp. The porphyrin excretion pattern of PCT has three main features: (1) increased urinary excretion of URO and of other acetate-substituted porphyrins; (2) a distinctive pattern of excretion of isomer series I and III porphyrins; and (3) increased excretion of fecal ISOCOPRO. PCT patients excrete greatly increased amounts of urinary 8-carboxyl URO and also porphyrins with 7, 6-, and 5-carboxyl groups; the level of 4-carboxyl porphyrin (COPRO) is also increased but to a lesser extent than that of URO and rarely surpasses 600 µg per 24 hours (see Table 132-7). In PCT, the hepatic UROGEN decarboxylase deficiency results in the accumulation of 5-carboxyl porphyrinogen III . This can be used as a substrate by the enzyme COPROGEN oxidase, thereby forming dehydroisocoproporphyrinogen, which in turn is oxidized to ISOCOPRO. This results in the characteristic elevation of this compound in the feces of these patients. The 8-carboxyl URO and 7-carboxyl porphyrins are the predominant urinary porphyrins in patients with PCT (more than 90 percent of total porphyrins). The urinary porphyrin excretion pattern is a mixture of type I and type III isomers. URO is approximately 60 percent type I isomer and 40 percent type III; the 7- and 6-carboxyl porphyrins are more than 90 percent type III and less than 10 percent type I isomer; the 5- and 4-carboxyl porphyrins are approximately 50 percent each isomer. This distinctive isomer pattern is found consistently in patients with PCT. In general, only trace amounts of URO are present in the stool of individuals without porphyria. The porphyrin content of stool is increased in patients with PCT and consists primarily of ISOCOPRO (type III), 7-carboxyl porphyrin, and lesser amounts of URO and COPRO. The total daily 24-hour fecal porphyrin excretion may exceed total urinary porphyrin excretion. The ratio of URO to COPRO in the urine is often helpful in differentiating PCT and VP. In PCT, the URO-COPRO ratio is usually greater than 3:1, whereas in VP the ratio is usually less than 1:1. Occasionally, 24-hour urine porphyrin levels will be normal or only slightly increased in a patient with the cutaneous findings of PCT. This should alert the physician to evaluate stool porphyrins, because these are elevated in patients with VP (see Variegate Porphyria). It should be emphasized that patients with clinical signs of PCT may have negative results on a urine fluorescent screening test for porphyrins. In such patients, it is absolutely essential to obtain plasma porphyrin levels and perform quantitative 24-hour urine URO and COPRO determinations and stool PROTO and COPRO determinations, preferably using high-performance liquid chromatography, which often permit differentiation of PCT from VP. Rarely, some patients with VP will have normal fecal porphyrin excretion, and bile porphyrin measurements may be helpful in evaluating such patients.100 Virtually all patients with PCT have excessive total body iron stores manifested as increased serum iron, ferritin, and/or hepatocellular iron levels. Occasionally, there is mild erythrocytosis. An abnormal glucose tolerance test result is seen in a minority of patients. Biochemical tests for liver function may be performed to identify liver disease. Elevated serum transaminase and γ-glutamyltranspeptidase levels may be found. Serum iron and ferritin concentrations may be elevated. The measurement of erythrocyte UROGEN decarboxylase is useful for the detection of patients with type II PCT. Mutational analysis is an essential part of evaluating these patients (see Molecular Genetic Research Strategies for the Porphyrias). HISTOPATHOLOGY The characteristic histopathologic finding in PCT is a sub-epidermal blister . Bullae typically show a corrugated, undulating base that has been termed festooned.101 There is little or no inflammatory infiltrate. Periodic acid-Schiff (PAS) stain reveals a mild degree of thickening of the papillary vessel wall, not nearly as marked as that seen in patients with EPP. Reticulin staining demonstrates slight proliferation of fibers along the basement membrane. Direct immunofluorescence studies reveal deposition of C3 and immunoglobulin G in a granular pattern at the dermal-epidermal junction and in and around vessel walls in affected individuals.102 These changes are most apparent in sun-exposed areas in patients with active disease and high urinary porphyrin excretion, and they decrease substantially in patients after appropriate treatment. It is also possible that the deposition of immunoglobulins and complement is a non-specific result of injury to the cutaneous tissue. The injury seen to the upper dermal vessels and at the dermal-epidermal junction suggests that damage to these areas induced by porphyrin photosensitivity may be responsible for the unique skin fragility seen in PCT. Regardless of these findings, it should be emphasized that histopathologic examination does not substantially contribute to confirming the diagnosis of PCT. This is also true for the other cutaneous porphyrias. Consequently, it is not essential to perform a skin biopsy if one of the cutaneous porphyrias is suspected, mainly for two reasons: First, simple non-invasive biochemical laboratory techniques (see Laboratory Tests earlier) can usually permit presumptive diagnosis of porphyria, and second, external trauma (such as a biopsy or excision) inevitably constitutes an avoidable risk for delayed and/or dysfunctional wound healing, which is a characteristic feature of all cutaneous porphyrias.
DIFFERENTIAL DIAGNOSIS Other dermatoses that can be confused with PCT include VP, HCP, mild forms of HEP and/or CEP, pseudoporphyria, scleroderma, and epidermolysis bullosa acquisita. Each of these can be differentiated by appropriate porphyrin studies. Careful evaluation of urine, stool, and plasma porphyrins will almost always permit confirmation of the diagnosis of PCT. Nonsteroidal anti-inflammatory drugs, such as naproxen, and antibiotics, such as the tetracyclines and nalidixic acid, as well as a variety of other agents, may rarely produce bullous lesions closely resembling PCT but in contrast to it show no evidence of abnormal porphyrins (see Pseudoporphyria). TREATMENT Initially, a careful history should be taken in an effort to identify an environmental toxin (e.g., alcohol, estrogen, or chlorinated hydrocarbon) that may have triggered the disease. If HCV or HIV infection is present, these should be managed appropriately. Elimination of toxin exposure alone may result in gradual improvement. In most patients with PCT, however, more aggressive treatment is usually appropriate to accelerate therapeutic benefit, and this currently consists of either repeated phlebotomy or orally administered antimalarials (either chloroquine or hydroxychloroquine), or a combination of both.107 Other forms of treatment that have been described include administration of iron chelators and the oral administration of cholestyramine. Phlebotomy. Phlebotomy is still the treatment of choice for PCT. Numerous reports have emphasized the safety and efficacy of this form of therapy, which was introduced by Ippen. Phlebotomy is effective because it depletes the excessive hepatic iron stores characteristic of PCT. Biochemical remission of PCT has occurred in patients treated with phlebotomy with iron overload as well as in patients with quantitatively normal iron stores. Replenishment of iron after phlebotomyinduced remission of PCT has resulted in biochemical and clinical exacerbation of the disease. Abstinence from the environmental triggers alone, especially alcohol, may induce a clinical and biochemical remission, although this may take months to years. Treatment of the Non-Acute Porphyrias
The efficacy of phlebotomy may relate to several effects:
Phlebotomy is carried out as an ambulatory procedure. The total amount of blood removed varies widely, usually ranging from 1500 to 12,000 mL. It is most convenient to use plastic blooddrawing bags available in any blood bank. Approximately 500 mL of blood is removed at weekly or biweekly intervals until the hemoglobin level decreases to approximately 10 g/dL or until the serum iron level drops to 50 to 60 µg/dL. Some believe that phlebotomies should be continued until serum ferritin level falls to the lower range of normal. Patients are strongly encouraged to discontinue or decrease exposure to porphyrinogenic agents, because this usually hastens clinical and biochemical remission. It is particularly important to reassure the patient that clinical improvement may not become apparent for variable intervals after the phlebotomies are begun. Porphyrin excretion continues to fall long after phlebotomies are discontinued. It has been shown that in more than 90 percent of patients treated with regular phlebotomy, urinary URO excretion reaches normal levels (less than 100 µg/24 hour) after 5 to 12 months. Blistering is the first sign to disappear; this is followed by improvement in skin fragility and in hypertrichosis over a period of 3 to 18 months. Even sclerodermoid changes can resolve slowly, although this may take several years. There are few published data on long-term follow-up of treatment, but most patients who experience relapse have again responded to repeated courses of phlebotomy.104 The length of remission induced by phlebotomy varies widely and ranges from 6 months to more than 10 years. At least 10 percent to 20 percent of patients will experience relapse within 1 year. Phlebotomy is a safe, effective, and relatively simple form of therapy with minimum associated morbidity. A few patients may complain of mild to moderate fatigue and weakness during the treatment period, but this usually resolves as the hemoglobin level returns to normal. Antimalarials. In some patients, phlebotomy is not recommended or is contraindicated because of the presence of anemia, cardiopulmonary disorders, or HIV infection. In such cases low-dose antimalarial therapy is a useful alternative. The antimalarial aminoquinolines chloroquine and hydroxychloroquine are used. In 1957, it was first suggested that chloroquine was useful in treating PCT. The cutaneous signs of the disease cleared within 1 year in one patient who received 500 mg daily for several months. Such dosages may trigger severe hepatotoxicity in many PCT patients, however, and this is no longer considered an acceptable approach. Instead, low-dose chloroquine therapy at a dosage of 125 mg twice weekly is effective in treating PCT and avoids the hepatotoxic effects of high-dose therapy. In addition to producing a successful therapeutic outcome in single patients and small cohorts, low-dose chloroquine (125 mg twice weekly for 8 to 18 months) has been used successfully to treat more than 100 patients with PCT.116 Liver function test results and urinary porphyrin levels are monitored quarterly, and the medication is continued until urinary URO level is less than 100 µg/24 hours. This usually requires 6 to 12 months of treatment. Studies comparing the therapeutic efficacy of phlebotomy with that of low-dose chloroquine suggest that they are equally efficacious, and, likewise, the clinical and biochemical remission of PCT obtained with chloroquine appears to be identical in all respects to that induced by phlebotomy. However, it has been reported that rapid relapse occurred in several patients treated with hydroxychloroquine, and therefore chloroquine may be preferable. The mechanism of action of chloroquine may relate to the formation of water-soluble drug-porphyrin complexes that are then excreted from the liver.118 Taljaard et al., however, believe that chloroquine chelates iron in the hepatocyte and that the bound iron is then eliminated. The combination of phlebotomy and chloroquine treatment may reduce the severity of the hepatotoxic response to chloroquine and also accelerate remission of the disease.107 Patients are treated with a series of one to four phlebotomies before starting chloroquine therapy (250 mg daily for 7 days). The procedure is repeated when signs of biochemical or clinical relapse develop, which may occur in 1 to 2 years. It should be emphasized that despite the tendency of the antimalarials to evoke hepatotoxic responses in patients with PCT, there is no evidence to suggest that the changes in hepatocellular pathology characteristic of PCT worsen as a result of treatment with these drugs.The antimalarials may cause retinopathy, and the low-dose regimen helps to minimize the risk of this complication. Pre-treatment and semi-annual ophthalmologic examinations should be performed in patients treated with these drugs. In patients with PCT and chronic renal failure, standard phlebotomy and antimalarial therapy are not feasible. The combination of high-dose recombinant erythropoietin and small-volume phlebotomy has been successful in selected patients. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||