| Superficial morphea = القشيعة السطحية |
|
|
Morphea MORPHEA Localized scleroderma (LS), or morphea, differs from systemic sclerosis (scleroderma) by the presence of various cutaneous morphologic variants and the absence of clinically detectable systemic involvement. There may be overlaps in the pathogenesis of fibrosis in the localized and systemic forms. Epidemiology LS has been reported to have an incidence of 2.7 per 100,000. There is a male-female ratio of 1:2 to 3, and the condition is more common in Caucasians and Asians than in African Americans. All variants occur in adults and children and can occur at any age. Linear scleroderma is more common in children and can present in the first or second decade, whereas morphea and generalized morphea are more common in adults and usually present in midlife. The relative frequency of the different morphologic variants is not clear, and reported studies have found different rates. In affected adults, 35 percent to 65 percent have plaque-type morphea, 8 percent to 9 percent generalized morphea, 6 percent to 46 percent linear scleroderma, 3.5 percent en coup de sabre, 3.5 percent morphea profunda, and less than 1 percent guttate, bullous morphea, and Parry-Romberg syndrome. One study, which included 126 children, found that 48 percent of patients had plaque-type morphea, 17.5 percent linear, 14 percent mixed morphologic type, 8 percent Parry-Romberg, 3 percent generalized morphea, 3 percent en coup de sabre, 2 percent a keloidal form, and less than 1 percent had pansclerotic morphea of childhood, morphea profunda, or bullous morphea. The average duration of clinically active disease ranges from 3 to 6 years. In chronic cases, slow clinical progression may persist for decades. Most patients will have a clinical remission of disease. Patients with plaque-type morphea are most likely to have the shortest disease duration. Rare patients, especially those with generalized morphea, morphea profunda, or linear scleroderma, will have a chronic course with several relapses, sometimes barely detectable clinically. Etiology and Pathogenesis The etiology of LS is unknown. There are reports of morphea after infection with measles, varicella, and Borrelia burgdorferi. Other suggested triggers include trauma, bacille Calmette-Guérin vaccination, vitamin B injection, radiation therapy, penicillamine, and bromocriptine. However, no direct etiology has ever been proved. Suggested pathogenic events are similar to those hypothesized for systemic sclerosis. Endothelial cell damage, inflammation and release of cytokines that stimulate collagen production by fibroblasts, and an imbalance of extracellular matrix turnover in the skin seem likely. The reason for the limited distribution of LS is unclear, but the patterning suggests that mosaic genetic changes may contribute . Many studies have suggested a pathogenic role for transforming growth factor-β (TGF-β). TGF-β stimulates fibroblasts to produce increased amounts of glycosaminoglycans, fibronectin, and collagen; decreases extracellular matrix breakdown; and it diminishes fibroblast susceptibility to apoptosis. TGF-β has been found to be increased in lesions of LS as well as in the skin and fibrotic lungs of patients with systemic sclerosis. Some work suggests that, at least in systemic sclerosis, TGF-β receptor expression in dermal fibroblasts is increased. There are also data supporting the possibility that alterations in the Smad protein pathway, which is important in TGF-β signal transduction, may play a role in collagen overproduction.12-18 Fibroblast cultures derived from systemic sclerosis and LS produce increased amounts of connective tissue components, including collagen type I in vitro. Skin biopsies have shown a greater capacity for collagen overproduction and sub-population of fibroblasts with activation of type I collagen expression; these fibroblasts co-localize with inflammatory mononuclear cells that express TGF-β. Biopsies of sclerotic lesions also show expression of different isoforms of TGF-β, as well as tissue metalloproteinase-3 (TIMP-3) in sub-populations of fibroblasts cultured from LS lesions. TGF-β enhances TIMP-3 expression, and TIMP-3 inhibits breakdown of collagen.19 It should be noted that three different isoforms of TGF-β (1, 2, and 3) are present in humans; TGF-β1 is the best studied. Some evidence suggests that the fibrotic response may be driven predominantly by CD4+ T cells. Plasma cells and histiocytes can probably contribute to the stimulation of dermal fibroblasts. Inflammatory cells found in the dermis of scleroderma lesions are primarily T lymphocytes, mainly T helper cells. There is also increased interleukin 2 (IL-2) and IL-4 production.20 At least in the systemic form of scleroderma, connective tissue growth factor has also been implicated.11,21,22 A pathogenic role for dermal dendrocytes has also been suggested. The presence of CD34+ and of factor XIIIa+ dermal dendrocytes correlates with active inflammation and sclerosis in LS.9 The pathogenic role of mast cells in LS has not been clearly elucidated, but mast cells may be a component of sclerodermatous skin, especially in the inflammatory and early stages. Mast cell granules contain chemical mediators and proteolytic enzymes that can stimulate fibroblasts and even activate profibrotic cytokines, (i.e., TGF-β); histamine may also stimulate collagen production. Clinical Findings There are three main variants of LS: morphea , generalized morphea , and linear scleroderma . Morphea and generalized morphea have a slow and insidious onset and typically affect the trunk. Other less frequent variants include bullous, keloidal, guttate, and subcutaneous morphea . Linear scleroderma is more common in children younger than
CUTANEOUS LESIONS Morphea. Morphea presents with a single or a few circumscribed indurated patches or plaques generally with hypo- or hyperpigmentation . Early lesions present with edema with or without surrounding erythema. Pain occasionally is present for weeks before becoming clinically apparent. Active lesions can become indurated and maintain an erythematous or violaceous border. With progression, lesions often become whitened or yellow , especially centrally. Plaques vary in size from 0.5 to 30 cm.2 Sub-morphologic variants of morphea include guttate, bullous, keloidal, and morphea profunda . Some authors suggest that lichen sclerosus is a superficial or early form of morphea ; indeed, the white, atrophic lesions of morphea may resemble lichen sclerosus. Chronic sclerodermoid graft-versus-host disease can have clinically and histologically overlapping features of lichen sclerosus, morphea, and eosinophilic fasciitis (EF), suggesting that these disorders may be part of a spectrum of fibrotic disease.
Generalized Morphea. Generalized morphea is a more severe form of morphea characterized by multiple lesions, often showing confluence and involving a larger body surface area . Some patients may have a subcutaneous form with little surface involvement . Linear Scleroderma. Linear scleroderma is characterized by a band-like skin induration often with pigment changes, which may cross joint lines and sometimes leads to contractures . This form of LS occurs more commonly in children and on the extremities. The fibrotic process often may extend to the subcutaneous tissue, including fascia and muscle . Joint contractures can be a significant cause of morbidity and deformities . In very young children, the process may affect bone growth and disrupt development of tissues. A “pansclerotic” process involving the entire extremity is seen in very severe cases. Pansclerotic morphea in children has been associated with an increased risk of cutaneous squamous cell carcinoma, particularly in ulcerated areas of affected skin.29,30 Linear scleroderma occurring on the face may present either as a purplishbrown streak or as a single white, atrophic band running vertically on the forehead, generally known as en coup de sabre . Progression to involve the scalp is common . If only the subcutaneous tissue, muscle, and, occasionally, bone are involved, this ipsilateral form
Eosinophilic Fasciitis. EF, or Shulman syndrome, is a related disorder that presents with the rapid onset of symmetric areas of edema on the extremities . The condition progresses over several weeks to cutaneous induration and contractures, with features similar to those of subcutaneous forms of LS.2 Up to 30 percent to 40 percent of patients with EF have lesions indistinguishable from LS. Atrophoderma of Pasini and Pierini. Atrophoderma of Pasini and Pierini is present in many patients with LS and presents with oval blue to dark brown hyperpigmented atrophic and slightly depressed areas. There is generally no induration or violaceous border in these lesions, in contrast to plaque-type morphea. The borders of the atrophoderma lesions have a “cliff-drop” appearance . Some consider atrophoderma to be an aborted, superficial form of LS . RELATED PHYSICAL FINDINGS Typically, LS patients have no signs beyond the cutaneous lesions. Initially, before the development of LS, patients may present with arthralgia and, occasionally, frank arthritis. These findings tend to resolve with progression of the cutaneous lesions, but may cause clinicians to consider systemic rheumatologic conditions. Other uncommon findings include fever, regional lymphadenopathy, migraines, abdominal pain, tinnitus, neuralgias, and spina bifida.2,4 En coup de sabre has been associated with neurologic abnormalities, including seizures, uveitis, and inflammatory lesions of cortical and subcortical regions of the brain. However, there is no recognized internal organ involvement in LS. LABORATORY ABNORMALITIES Serum Autoantibodies. Serum autoantibodies have been found with variable frequency in patients with LS. The most commonly found autoantibodies are antinuclear antibodies in up to 46 percent to 80 percent of patients,5,6,33,34 usually with a homogenous immunofluorescence pattern. With extensive involvement, 36 percent to 53 percent of cases have anti-single stranded DNA and/or antihistone antibodies.33,35,36 Typically, patients with generalized morphea have a higher frequency of antibody positivity than other subsets of LS, and autoantibodies correlate with a more severe clinical presentation, greater number of lesions, more sclerotic lesions, and a longer duration of clinical course. Other Serum Abnormalities. Blood eosinophilia has been described in 6 percent to 50 percent of patients with LS. Levels of eosinophilia appear to correlate with disease activity. Declining levels of eosinophilia may coincide with a decrease in activity of the cutaneous lesions. Elevated immunoglobulins, particularly serum immunoglobulin G levels, have been associated with active, more extensive disease and joint contractures. A positive rheumatoid factor is seen in 26 percent of patients, and the erythrocyte sedimentation rate is elevated in 25 percent.
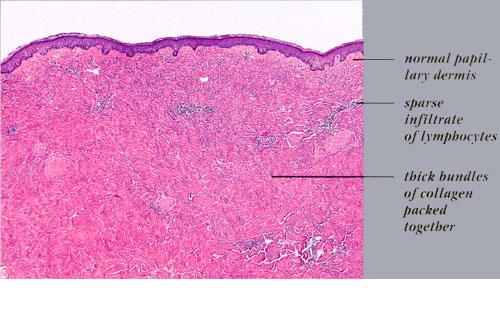
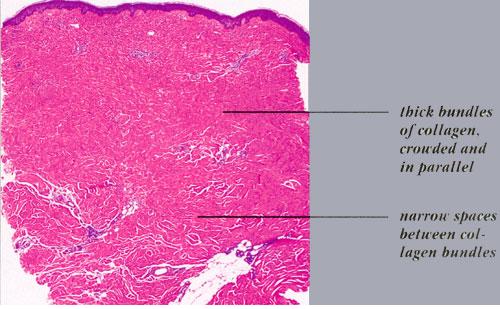
Histopathology. Early lesions may not have obvious or specific histologic changes. Vacuolization and destruction of endothelial cells with basal lamina reduplication have been reported.2 Typically, in advanced indurated lesions, one sees a squared-off edge to the biopsy specimen . Superficial and deep inflammatory infiltrates may occasionally be present. Very early lesions may demonstrate an inflammatory infiltrate in the deep dermis and subcutaneous tissue (see eFig 62-6.1 in on-line edition). Lymphocytes, macrophages, plasma cells, eosinophils, and mast cells are also seen. Deposition of glycosaminoglycans may be detected in the early stages of LS, particularly if care is taken to preserve this matrix component during histologic processing.
Differential Diagnosis Complications Subcutaneous and muscle tissue atrophy and joint contractures are mainly seen in linear scleroderma , generalized morphea , and subcutaneous morphea (morphea profunda), and can cause significant impairment of mobility. Contractures are frequently seen in linear scleroderma involving limbs and crossing joint lines . Children are more commonly affected by linear scleroderma than adults. In rare and severe cases, pansclerotic morphea may require amputation of the involved limbs because of their impaired growth. Patients with linear craniofacial involvement , such as en coup de sabre and facial hemiatrophy, may have neurologic, ophthalmologic, and oral abnormalities. Severe cases of LS characterized by hyper- or hypopigmentation, contractures, and underlying tissue atrophy can be devastating. These complications lead to functional, cosmetic, and psychological difficulties.2,5 Differential Diagnosis of Morphea Most Likely
Consider
Always Rule Out
POEMS = polyneuropathy, organomegaly, endocrinopathy, M protein, and skin changes. Prognosis and Clinical Course In spite of the common presence of serum autoantibodies, LS typically does not have a recognized systemic involvement, although a rare overlap with other connective tissue disorders has been reported. Most cases of LS are self-limited, with clinical activity apparent for an average of 3 to 5 years. Some patients may have re-activation of apparently inactive lesions. In up to 13 percent of patients with linear scleroderma, one sees re-activation after several years of remission. En coup de sabre may have an insidious course lasting for decades. It may be that LS remains a chronic process with a low level of activity over many years. Slight atrophy with or without hyperpigmentation may be the only persistent sign of disease. Treatment In many cases, LS lesions become inactive spontaneously; however, more severe cases can cause irreversible fibrosis/sclerosis of the skin and subcutaneous tissues. Treatment is directed at the inflammatory component, cytokine release, and activation and collagen deposition. Many therapies have been used in the treatment of LS with variable success. Topical and systemic steroids, oral and topical vitamin D analogues, methotrexate, cyclophosphamide, azathioprine, hydroxychloroquine, intralesional interferon-γ, penicillin and D-penicillamine have all been tried. Physical therapy is of the greatest importance in patients with contractures to preserve or improve limb function. Treatments that have been reported to be successful include D-penicillamine, topical tacrolimus under occlusion, oral calcitriol, topical calcipotriene, methotrexate alone42 or combined with pulsed corticosteroids, topical imiquimod, topical tretinoin with ammonium lactate, and N-(3′, 4-dimethoxycinnamoyl) anthralinic acid—an antiallergy medication that inhibits passive cutaneous anaphylaxis. In pediatric cases with impaired growth of the affected extremity, timely surgical intervention and “stapling” of the epiphyseal plates of the normal side may be effective. This will result in slower, but continued, growth of the affected extremity and may lead to a lesser degree of limb disparity. PHOTOTHERAPY Several studies have demonstrated marked improvement in a majority of LS patients using psoralen and ultraviolet A light, broad band ultraviolet A (UVA), or UVAI phototherapy. PRACTICAL APPROACH In the absence of truly randomized and prospective clinical trials, the approach to patients is often highly individualized. For limited involvement with one or few morphea lesions, one can use topical treatments such as calcipotriene, tacrolimus, retinoids, or no treatment at all. On the other hand, lesions of en coup de sabre can lead to considerable disfigurement. Our approach to those facial lesions has been the use of either hydroxychloroquine or perhaps methotrexate in combination with small doses (5 to 10 mg) of systemic corticosteroids. For more generalized involvement, we usually use phototherapy. If that approach is not successful, or if there is substantial subcutaneous involvement, a useful medication is methotrexate. Beyond these approaches, one is left with even more anecdotal experience. D-penicillamine, cyclosporine, and other immunosuppressive agents have been used.
|