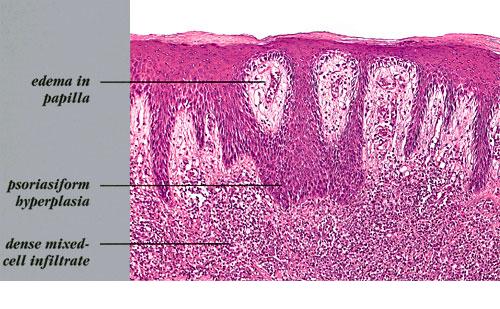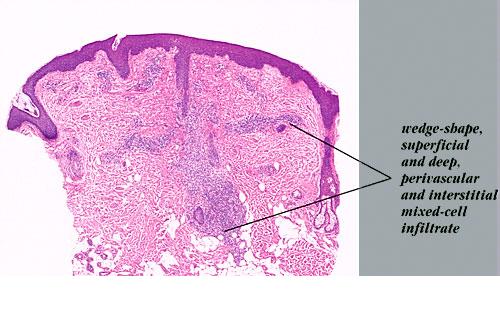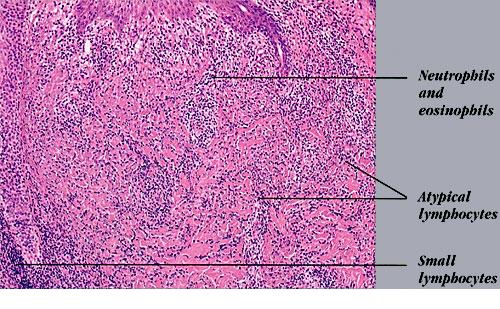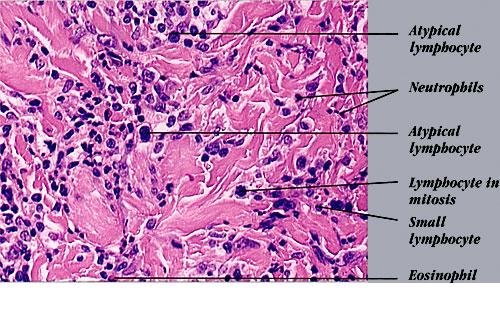









Lymphomatoid Papulosis
Lymphomatoid papulosis (LyP) is a chronic papulonecrotic or papulonodular skin disease with histologic features suggestive of a malignant lymphoma. The disease is characterized by recurrent crops of pruritic papules at different stages of development that predominantly arise on the trunk and limbs. The papules heal spontaneously over 1-2 months, usually leaving slightly depressed oval scars.
The term lymphomatoid papulosis originally was used by Macaulay1 in 1968 to describe "a self-healing rhythmical paradoxical eruption, histologically malignant but clinically benign." Due to the typical waxing and waning clinical course, lymphomatoid papulosis was previously considered a pseuodolymphomatous inflammatory process. However, the classification system for cutaneous lymphomas has evolved rapidly, and, during consensus meetings in 2003-2004, the World Health Organization—European Organization for Research and Treatment of Cancer (WHO-EORTC) classification grouped lymphomatoid papulosis among the indolent cutaneous T-cell lymphomas. The rationale for classifying lymphomatoid papulosis as a cutaneous lymphoma is its association with other malignant lymphoproliferative disorders; however, some experts hesitate to classify this chronic skin disease as a true malignancy because of its spontaneous resolution and benign clinical course.2,3,4
Lymphomatoid papulosis is part of a spectrum of CD30 (Ki-1)–positive cutaneous lymphoproliferative diseases (CD30+ LPDs), including lymphomatoid papulosis, primary cutaneous anaplastic large cell lymphoma (pcALCL), and borderline CD30+ lesions. Also see Cutaneous CD30+ (Ki-1) Anaplastic Large-Cell Lymphoma.
Also see the eMedicine articles Malignant Melanoma (dermatology focus), Malignant Melanoma (oncology focus), Cutaneous T-Cell Lymphoma, and Lymphoma, Cutaneous T-Cell.
Pathophysiology
The CD30 antigen is a type 1 transmembrane glycoprotein of the tumor necrosis factor receptor superfamily. In addition to the CD30+ lymphoproliferative diseases, malignant lymphomas such as Hodgkin disease (HD), node-based systemic anaplastic large cell lymphoma (ALCL), and mycosis fungoides (MF) with large cell transformation may express the CD30 antigen.
The pathophysiology of CD30+ LPDs, including lymphomatoid papulosis (LyP), is largely unknown. CD30 signaling is known to have an effect on the growth and survival of lymphoid cells, and one hypothesis is that genetic instability and accumulated genetic defects may have a role in the development of lymphomatoid papulosis and the progression to associated neoplasms. In a recent study, the 30M377 allelic form of the CD30 promoter microsatellite repressive element was associated with the development of lymphomatoid papulosis, and the 30M362 allelic form was associated with progression to other CD30+ lymphomas in lymphomatoid papulosis patients.
Genetic instability of tumor cells may lead to altered expression of apoptotic proteins and immune-regulatory molecules, such as transforming growth factor-beta. Other research has found overexpression of JunB,5 part of the AP-1 transcription factor complex involved in cell proliferation and apoptosis, in virtually all CD30+ lymphomas. Consequently, JunB is a potential target for experimental therapy in patients with these tumors.
Spontaneous regression of lymphomatoid papulosis is seen almost universally, whereas regression occurs in approximately 25% of pcALCL cases. Therefore, the higher apoptotic index found in lymphomatoid papulosis compared with pcALCL is not surprising. The proapoptotic protein Bax is also expressed at high levels in CD30+ cutaneous lymphoproliferative diseases and may play a crucial role in mediating apoptosis of tumor cells. Another recent study suggested that the death-receptor apoptosis pathway mediated by Fas-associating protein with death domain (FADD) may be responsible for the varying biologic behaviors of CD30+ LPDs involving the skin.6
The prevalence of lymphomatoid papulosis is estimated to be 1.2-1.9 cases per million population. The CD30+ cutaneous lymphoproliferative disorders account for approximately 25% of cutaneous T-cell lymphoma cases.
Lymphomatoid papulosis has a chronic, indolent course in most patients; however, estimates indicate that as many as 10-20% of lymphomatoid papulosis patients have a history of associated malignant lymphoma (ALCL, HD, or MF) prior to, concurrent with, or subsequent to the diagnosis of lymphomatoid papulosis. In a recent longitudinal study, no patients with lymphomatoid papulosis died of the disease.
pcALCL is more likely than MF to manifest as an ulcerated tumor and palpable lymph nodes. MF is the most common variant of cutaneous T-cell lymphoma and is characterized by the development of red patches or plaques in sun-protected areas. MF is more likely to manifest as patches and plaques than tumors. Disease-specific survival at 5 and 10 years for pcALCL was 85% in a recent study.
Associated lymphomas more rarely include immunoblastic lymphoma, lethal midline granuloma (currently considered as natural killer cell lymphoma in many patients), and systemic lymphocytic lymphoma. In most patients, the malignancy develops many years after the diagnosis of lymphomatoid papulosis.
Black persons may be less affected by lymphomatoid papulosis than persons of other racial groups.
No consistent sex predominance is found in studies of lymphomatoid papulosis, but some studies have reported a male-to-female ratio of 1.5-2:1.
Lymphomatoid papulosis may develop at any age, but the peak incidence occurs in the fifth decade.
History
- Most patients with lymphomatoid papulosis (LyP) describe the gradual onset of an asymptomatic to mildly pruritic papular eruption.
- Papules appear in crops and resolve spontaneously within 2-8 weeks.
- Waxing and waning of the crops of papules may continue for decades.
- Unless accompanied by systemic lymphoma, most patients have no constitutional symptoms.
Physical
- Unless accompanied by systemic lymphoma, physical findings are limited to the skin and, very rarely, the oral cavity.7,8
- The primary skin lesions are described as follows:
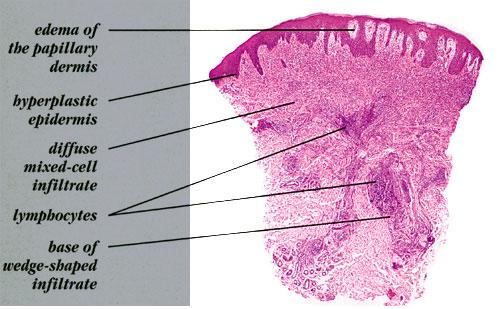
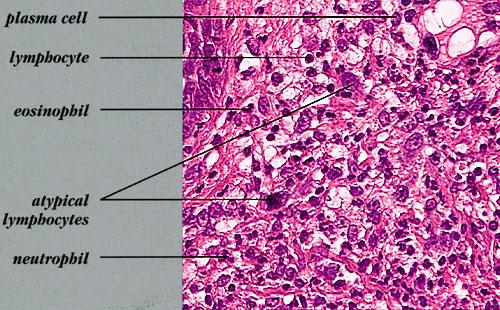
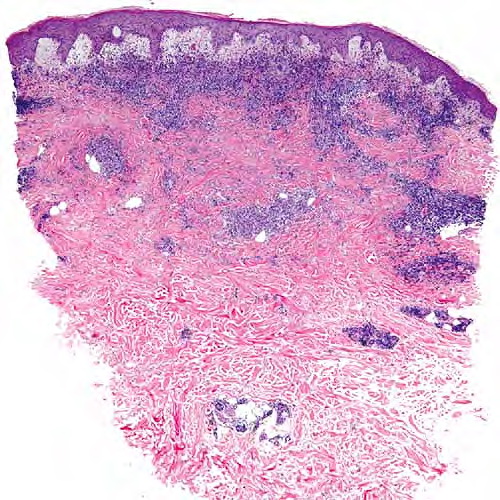
- Each erythematous papule evolves into a red-brown, often hemorrhagic, papulovesicular or papulopustular lesion over days to weeks, as demonstrated in the images below.
-
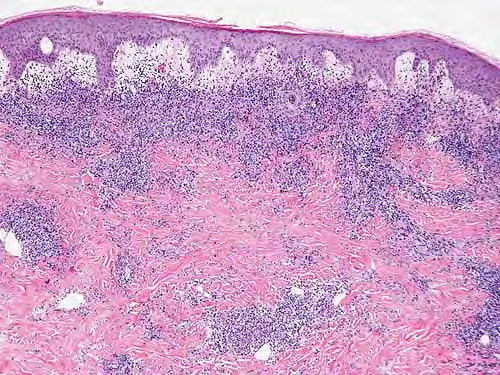
- Some lesions develop a necrotic eschar before healing spontaneously. Occasionally, noduloulcerative lesions may be present, as in the image below.
-
- Each papule heals within 2-8 weeks, leaving a hypopigmented or hyperpigmented, depressed, oval, and varioliform scar.
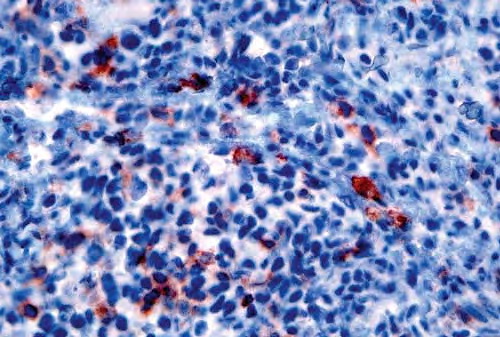
- Large nodules and plaques may take months to resolve. Carefully evaluate solitary ulcerated nodules, plaques, or masses for CD30+ ALCL (see image below), MF, or rarely, HD.
-
- The skin distribution of lesions, characteristically, is on the trunk and extremities, although the palms and/or soles, face, scalp, oral mucosa,9 and anogenital area also may be involved.
- Evolving lesions have been described under dermoscopy. The initial papular lesion showed a vascular pattern of tortuous vessels radiating from the center. A white structureless area was seen around the vessels. More mature lesions, hyperkeratotic papules, looked similar except the vascular pattern in the center of the lesion was obscured. As the lesions progressed to necrotic ulcerations, the vascular pattern was only seen at the periphery, while the center of the lesions was a structure of brownish-gray areas. The final, or cicatricial phase, was similar except no vessel pattern was seen.10
Causes
- The etiology of lymphomatoid papulosis is unknown. Debate persists over whether (1) lymphomatoid papulosis is a benign chronic disorder of activated T cells responding to external or internal stimuli or (2) lymphomatoid papulosis is an indolent T-cell malignancy localized to skin and held in check by the host immune system.
- A few investigators have discovered viruslike particles in lymphomatoid papulosis lesions examined under electron microscopy.11
Treatment
Medical Care
- In the past, localized mildly pruritic skin lesions were treated with mid- to high-potency topical steroids to hasten resolution. Many authorities currently are more inclined to treat lesions with systemic or more aggressive topical therapies, including phototherapy, to suppress the disease and the possibility of progression to Hodgkin disease (HD), anaplastic large cell lymphoma (ALCL), or mycosis fungoides (MF).
- Low-dose weekly methotrexate (MTX) is a safe and effective treatment for suppressing lymphomatoid papulosis (LyP); however, the disease recurs within 1-2 weeks after discontinuing the medication.
- Oral psoralen plus UVA (PUVA) phototherapy also effectively treats and suppresses the disease.
- One report describes successful treatment of recalcitrant lymphoma papulosis in using PUVA-bath photochemotherapy in a pediatric patient.21
- A few reports also have found that topical carmustine, topical nitrogen mustard, topical MTX, topical imiquimod cream,22 intralesional interferon, low-dose cyclophosphamide, chlorambucil, medium-dose UVA-1 therapy, excimer laser therapy,23 photodynamic therapy,24 and dapsone help disease suppression.
Consultations
- Dermatologist: Consultation is recommended for evaluating clinical findings and obtaining skin biopsy specimens of appropriate lesions. Ideally, consult a dermatologist with experience in the management of cutaneous lymphomas.
- Dermatopathologist: Consultation is recommended for histologic evaluation of skin biopsy specimens, with occasional consultation by a hematopathologist for patients with borderline biopsy results.
Activity
Lymphomatoid papulosis mandates no activity restrictions.
Medication
The goals of pharmacotherapy are to reduce morbidity and to prevent complications.
Antimetabolites
Inhibit cell growth and proliferation by blocking key steps of the cell cycle.
Methotrexate (Folex, Rheumatrex)
Antimetabolite that inhibits DNA synthesis and cell reproduction in malignant cells; may suppress immune system. First-line oral agent for treatment of LyP. Disease usually is sensitive to low, weekly oral doses. Adjust dose gradually to attain satisfactory response.
Adult
5-10 mg PO qwk; may require up to 25 mg/wk to respond
Pediatric
Not established; 2.5-5 mg PO qwk suggested
Oral aminoglycosides may decrease absorption and blood levels of concurrent oral MTX; charcoal lowers levels; coadministration with etretinate may increase hepatotoxicity; folic acid or derivatives contained in some vitamins may decrease response to MTX; coadministration with NSAIDs may be fatal; indomethacin and phenylbutazone can increase plasma levels; may decrease phenytoin serum levels; probenecid, salicylates, procarbazine, and sulfonamides (including TMP-SMZ) may increase effects and toxicity of MTX; may increase plasma levels of thiopurines
Documented hypersensitivity; alcoholism; hepatic insufficiency; immunodeficiency syndromes; preexisting blood dyscrasias (eg, bone marrow hypoplasia, leukopenia, thrombocytopenia, significant anemia); breastfeeding
Pregnancy
D - Fetal risk shown in humans; use only if benefits outweigh risk to fetus
Precautions
Monitor CBC counts monthly and liver and renal function q1-3mo during therapy (monitor more frequently during initial dosing, dose adjustments, or when risk exists of elevated MTX levels, eg, dehydration); has toxic effects on hematologic, renal, GI, pulmonary, and neurologic systems; discontinue if significant drop in blood cell counts occurs; aspirin, NSAIDs, or low-dose steroids may be administered concomitantly (possibility of increased toxicity with NSAIDs, including salicylates, has not been tested); may cause mucositis or cutaneous ulceration
Phototherapy
Induce apoptosis in activated T cells. PUVA phototherapy effectively treats and suppresses disease.
Methoxsalen (8-MOP, Oxsoralen)
Inhibits mitosis by binding covalently to pyrimidine bases in DNA when photoactivated by UVA. In Europe, this modality is more popular than MTX for treating LyP. Available in 10-mg cap.
Adult
0.4 mg/kg/dose PO 1.5 h prior to UVA exposure; alternatively, 0.57 mg/kg 1.5-2 h before exposure to UVA, at least 48 h apart
Pediatric
Administer as in adults
Toxicity increases with phenothiazines, griseofulvin, nalidixic acid, tetracyclines, thiazides, and sulfanilamide
Documented hypersensitivity; squamous cell cancer; cataract; light-sensitive diseases such as lupus or porphyria; ingestion of photosensitizing drugs; hepatitic disease; arsenic therapy; history of melanoma; patients with aphakia
Pregnancy
C - Fetal risk revealed in studies in animals but not established or not studied in humans; may use if benefits outweigh risk to fetus
Precautions
Severe burns may occur from sunlight or UVA if dose or treatment frequency exceeded; use only if response to other forms of therapy is inadequate; long-term use may increase risk of skin cancer; ocular changes may occur