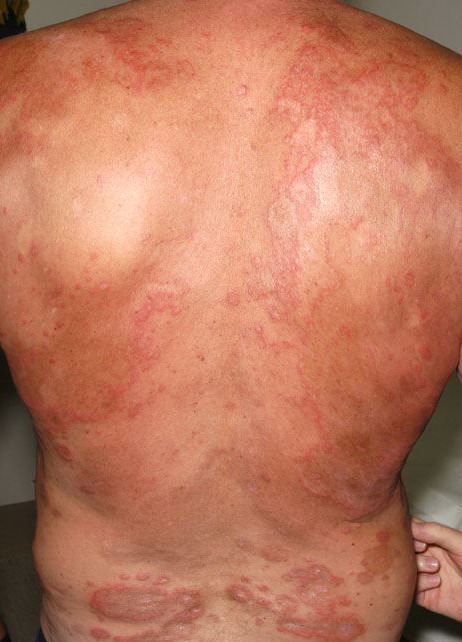
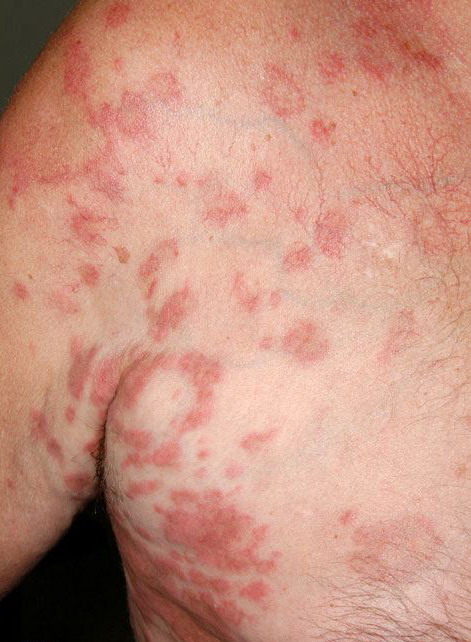
| Hypereosinophilic dermatitis= التهاب الجلد بفرط الحمضات |
|
|
HYPEREOSINOPHILIC
SYNDROMES
Epidemiology
The hypereosinophilic syndromes (HES) occur worldwide and span all age groups. Over 90 percent of patients with myeloproliferative HES and the mutant gene Fip1-like 1/platelet-derived growth factor receptor-α (FIP1L1-PDGFRA) are men, but lymphocytic HES shows equal gender distribution. The relative frequencies of the sub-types are unknown, although up to 25 percent of HES patients may have lymphocytic HES. Rare familial cases have been reported. A mini-epidemic has been observed over the last decade in eosinophilic esophagitis, a subset of overlap HES with organrestricted disease, with prevalence estimates as high as 1:2500 among children and 1:4000 among adults. Etiology Eosinophils are implicated as the cause of most end-organ damage in all HES subtypes. Clinical improvement usually parallels a decrease in eosinophil count. Patients with lymphocytic HES have abnormal T-cell clones with unusual surface phenotypes, including CD3+CD4-CD8- and CD3-CD4+. These T cells display activation markers, such as CD25, and secrete T helper 2 cytokines, including high levels of interleukin 5 (IL-5). An 800-kilobase deletion on chromosome band 4q12 that codes for a tyrosine kinase has been found in myeloproliferative HES. Patients with the FIP1L1-PDGFRA gene mutation, but without clinical manifestations of systemic mastocytosis, have elevated serum tryptase levels and increased atypical spindle-shaped mast cells in bone marrow. Although they do not exhibit all its immunologic markers, these patients satisfy criteria for mastocytosis but are considered to form a distinct subset of HES, with cardiomyopathy and endomyocardial fibrosis, that responds to imatinib. The FIP1L1-PDGFRA gene is detected in mast cells, eosinophils, neutrophils, and mononuclear cells. Many HES patients have marked neutrophilia, likely due to the aberrant gene in the neutrophil lineage. Thus, alteration of several cell lines probably contributes to the pathogenesis of myeloproliferative HES. Multiple other chromosomal abnormalities have been identified in myeloproliferative HES, including translocations, partial and complete chromosomal deletions, and trisomies 8, 15, and 21. The etiology of the other HES variants is not well understood, although patients with episodic angioedema and eosinophilia [Gleich syndrome; see Episodic Angioedema Associated with Eosinophilia (Gleich Syndrome)] and the nodules, eosinophilia, rheumatism, dermatitis, and swelling (NERDS) syndrome33 have developed T-cell clones. Clinical Findings and Course Patients satisfying HES diagnostic criteria present with signs and symptoms related to the organ systems infiltrated by eosinophils. HES may
EOSINOPHILS IN CUTANEOUS
DISEASES AT A GLANCE
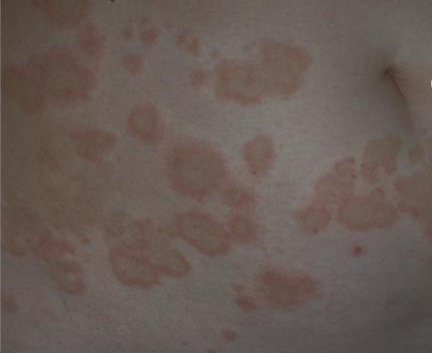
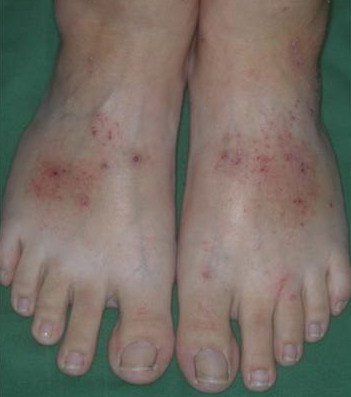
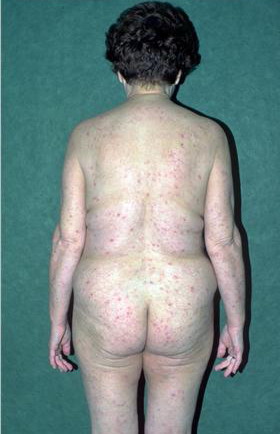
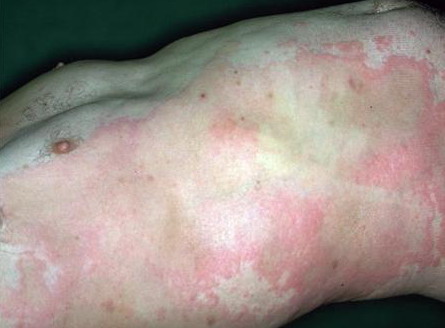
Lymphocytic HES commonly is associated with severe pruritus, eczema, erythroderma, urticaria, and angioedema, as well as lymphadenopathy and, rarely, endomyocardial fibrosis. In myeloproliferative HES, the usual presenting complex includes fever, weight loss, fatigue, malaise, skin lesions, and hepatosplenomegaly. Mucosal ulcers of the oropharynx or anogenital region portend an aggressive clinical course; death is likely within 2 years of presentation if the disorder is untreated. Cardiac disease occurs frequently. Eosinophils adhere to endocardium and release granule proteins onto endothelial cells, thrombus formation follows, and, finally, subendocardial fibrosis with restrictive cardiomyopathy occurs. Mitral or tricuspid valvular insufficiency results from tethering of chordae tendineae. Cardiac abnormalities that are essentially identical to those of HES but are confined to the intramural
HYPEREOSINOPHILIC SYNDROMES AT A GLANCE
Patients with myeloproliferative HES frequently present with clinical features resembling those of chronic myelogenous leukemia and sometimes are regarded as having chronic eosinophilic leukemia. Although the disease may evolve into frank leukemia, the relatively mature nature of the eosinophils and lack of evidence for clonal expansion generally preclude such classification. During the decade or more after diagnosis, HES may evolve into acute leukemia and, less commonly, has been associated with B-cell lymphomas. Churg-Strauss syndrome is a variant HES sub-type. Other variant HES types include Gleich syndrome [see Episodic Angioedema Associated with Eosinophilia (Gleich Syndrome)], in which eosinophil counts fluctuate with extreme angioedema. The overall 5-year survival rate for HES patients is 80 percent; congestive heart failure from the restrictive cardiomyopathy of eosinophilic endomyocardial disease is a major cause of death, followed by sepsis. Laboratory Tests A key criterion for diagnosis is marked, prolonged peripheral blood eosinophilia . Other causes of eosinophilia, including allergic and parasitic diseases, should be excluded. Tests to detect organ involvement, particularly measurement of liver enzyme levels, are important. Because eosinophilic endomyocardial disease can develop in any patient with prolonged peripheral blood eosinophilia, patients should undergo periodic echocardiography. Increased serum levels of immunoglobulin E (IgE) are often present in lymphoproliferative HES, and levels of vitamin B12 and tryptase are increased in myeloproliferative HES. The Chic2 fluorescent in situ hybridization assay detects the deletion that produces the FIP1L1-PDGFRA gene product and should be performed, because patients with this mutation respond to treatment with imatinib. Alternatively, the mutant gene can be detected by a polymerase chain reaction assay. Both tests are available commercially. In patients who lack the fusion gene, testing for other clonal cytogenetic abnormalities or abnormal clonal T-cell populations is warranted. Cytoflow of peripheral blood lymphocytes and immunophenotyping of tissue lymphocytes should be performed for the diagnosis of lymphocytic HES and repeated periodically to look for transformation from a variant HES type to lymphocytic HES or to T-cell lymphoma. The cutaneous histopathologic features of HES vary with the type of lesion. Skin biopsy specimens from urticarial lesions resemble idiopathic urticaria, with generally mild, non-specific perivascular and interstitial infiltration of lymphocytes, eosinophils, and, occasionally, neutrophils. Immunostaining reveals extensive deposition of eosinophil granule proteins in the absence of intact eosinophils in episodic angioedema with eosinophilia and HES with mucosal ulcers, and in synovial tissues in NERDS. Other than in Churg-Strauss syndrome, vasculitis is only rarely associated with HES. Differential Diagnosis Clinically, parasitic infections and infestations may closely resemble HES. A history of travel to endemic areas or certain dietary exposure implicates helminthiasis. Along with eosinophilia, total serum IgE levels higher than 500 IU/mL commonly are found in helminthic infections. Examination of stool samples for ova and parasites and serologic testing for Strongyloides antibodies should be performed. In patients with isolated urticarial plaques with or without angioedema, the differential diagnosis includes urticaria, but demonstration of multiorgan involvement supports HES. HES with episodic angioedema may resemble hereditary angioedema clinically, although patients with hereditary angioedema often have a family history of the disease, rarely have the markedly elevated eosinophil counts that characterize HES, and may be distinguished by complement abnormalities. Pruritic eczematoid lesions of lymphocytic HES may resemble those of atopic dermatitis, contact dermatitis, drug reaction,
Treatment
The goal of treatment is to relieve symptoms and improve organ function while keeping peripheral blood eosinophils at 1000 to 2000/µL and minimizing treatment side effects Myeloproliferative HES is very responsive to imatinib. In patients with the mutant gene FIP1L1-PDGFRA, administration of imatinib mesylate is indicated and usually induces hematologic remission, but endomyocardial disease may worsen during the first several days of treatment. Troponin levels should be monitored before and during imatinib therapy. To improve cardiac function, glucocorticoids should be given before and with initiation of imatinib therapy. Imatinib resistance can develop. In the absence of the gene mutation, after Strongyloides infection has been excluded, first-line therapy is prednisone. Approximately 70 percent of patients will respond, with peripheral eosinophil counts returning to normal. Patients for whom glucocorticoid monotherapy fails have a worse prognosis generally; in such cases, or when long-term side effects become problematic, other treatments should be used. Patients who have features of myeloproliferative HES but who lack FIP1L1-PDGFRA still may respond to imatinib. Interferon-α (IFN-α) has been beneficial in treating myeloid and lymphocytic HES. In one patient, loss of the FIP1L1-PDGFRA mutation after several years of IFN-α therapy was associated with complete remission. Other treatments with reported benefit include hydroxyurea, dapsone, vincristine sulfate, cyclophosphamide, methotrexate, 6-thioguanine, 2-chlorodeoxyadenosine and cytarabine combination therapy, pulsed chlorambucil, etoposide, cyclosporine, intravenous immunoglobulin, alemtuzumab, and psoralen plus ultraviolet A phototherapy. Extracorporeal photopheresis alone or in combination with IFN-α or other therapies, as well as bone marrow and peripheral blood stem cell allogeneic transplantation, represent additional options. Two monoclonal antibodies against human IL-5 have been associated with clinical improvement and reductions in peripheral blood and dermal eosinophils, particularly in patients with lymphocytic HES.
|