Congenital Hemangiomas
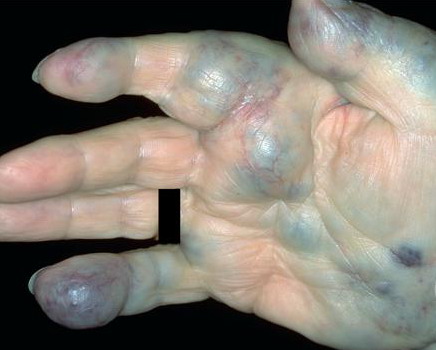
Hemangiomas that are fully formed tumors at the time of birth and do not proliferate in postnatal life are referred to as congenital hemangioma or congenital nonprogressive hemangioma. There are two major sub-types recognized on the basis of their natural history: the rapidly involuting congenital hemangioma (RICH) and non-involuting congenital hemangioma (NICH). Collectively, both are sometimes referred to as congenital nonprogressive hemangioma. The distinguishing pathologic features of these tumors are lobules of capillaries set within densely fibrotic stroma containing hemosiderin deposits, focal lobular thrombosis, and sclerosis.103 They are GLUT-1 negative. Both have similar anatomic sites of predilection, such as the extremities or postauricular skin, but they can occur elsewhere. RICH often appears as a raised, violaceous tumor
with large, radiating veins or with overlying telangiectasia and a halo of pallor . Central ulceration may be present. Most RICH involute spontaneously by age 14 months, often sooner, and usually leave residual atrophic inelastic skin in their wake.105
OTHER VASCULAR TUMORS AT A GLANCE
Congenital hemangiomas are fully formed at birth. Rapidly involuting congenital hemangiomas and non-involuting congenital hemangiomas are recognized sub-types.
· Tufted angiomas represent subtle pink or dusky-red patches and may evolve into plaques or nodules and have a characteristic histology.
· Kaposiform hemangioma is morphologically similar to but etiologically distinct from Kaposi sarcoma and can be associated with Kasabach-Merritt phenomenon.
· Multifocal lymphangiomatosis with thrombocytopenia consists of cutaneous vascular papules and plaques associated with intermittent thrombocytopenia, often with gastrointestinal bleeding.
· Spindle cell hemangioendothelioma usually occurs in the extremities most often associated with Maffucci syndrome.
· Congenital eccrine angiomatous hamartoma is a rare ill-defined plaque associated with increased lanugo hair and sweating.
· Pyogenic granuloma is very common; a rapidly growing papule or nodule with a collarette of scale or eroded surface. Treatment is excision or electrocautery.
NICH are also present at birth, but usually flatter than RICH, presenting as a well-circumscribed round to oval, slightly indurated or raised soft-tissue mass with overlying telangiectasias and a rim of pallor .
Both NICH and RICH are high-flow vascular anomalies, often showing arteriovenous micro-fistulas on Doppler interrogation. Some cases of RICH involute only partially, and the residual tumor resembles NICH, supporting the concept that RICH and NICH may be variants of each other.
Indications for treatment of NICH or RICH are similar to those for IH, including impairment of visual function and congestive heart failure. Excision should definitely be considered for ulceration, which in this setting can lead to severe hemorrhage, and for post-involutional skin changes if disfiguring. NICH do not go away but are often asymptomatic; decisions regarding their removal must weigh risks and benefits of the proposed treatment.
Tufted Angioma
ETIOLOGY
TA is a benign vascular tumor that has also been called angioblastoma of Nakagawa. Its etiology and pathogenesis are uncertain. Unlike IH, there are no known gender or gestational age correlates.
CLINICAL FINDINGS
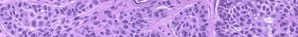
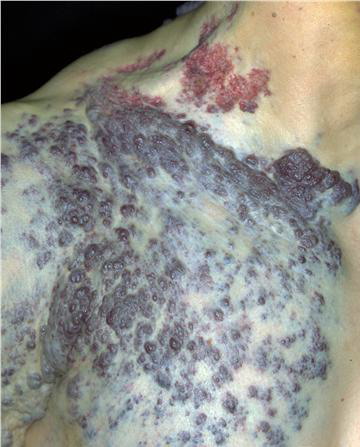
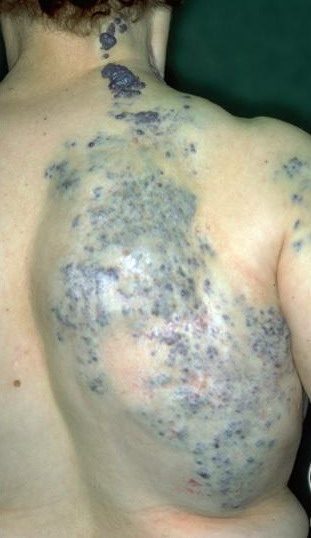
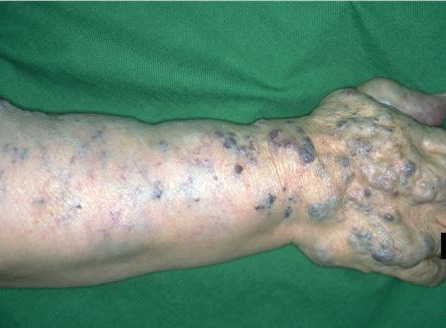
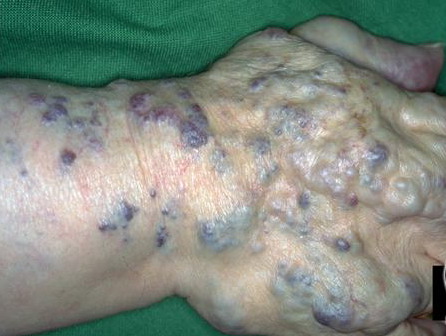
Most cases are acquired early in childhood and have a protracted course. Rare congenital forms also exist. TAs display various clinical patterns. They may present as a subtle stain-like area that later thickens, as a large, plaque-like, infiltrated, red or dusky blue-purple lesion, or as an exophytic, firm, violaceous, cutaneous nodule (Fig. 126-11). TA must be differentiated from infantile hemangiomas as well as other vascular tumors. They are often somewhat firmer and may be tender. Histologically, both acquired and congenital TAs demonstrate vascular tufts of tightly packed capillaries, randomly dispersed throughout the dermis in a typical “cannonball distribution” with crescentic spaces surrounding the vascular tufts, and lymphatic-like spaces within the tumor stroma.
COMPLICATIONS
KMP may develop in TA but this is more common with KHE (see the section Kaposiform Hemangioendothelioma).
PROGNOSIS AND CLINICAL COURSE
TA may persist unchanged or regress completely within a few years.
TREATMENT
No single treatment is effective. Interferon-α and PDL have anecdotally been reported to be effective