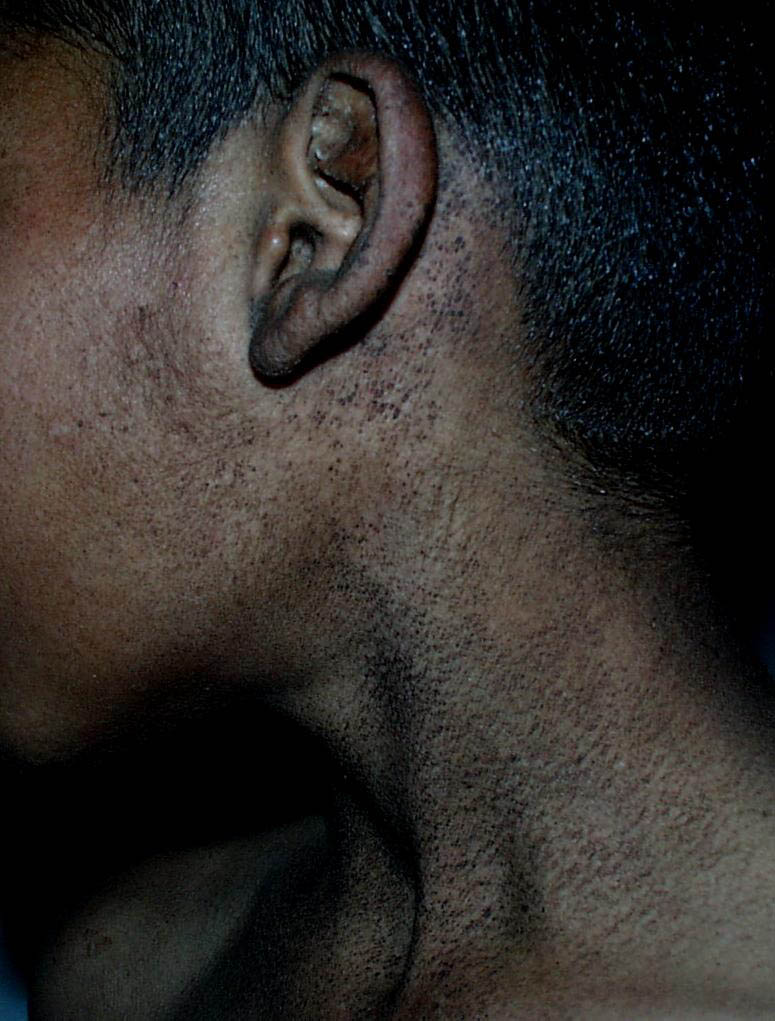▪ DARIER WHITE
DISEASE
Etiology and Pathogenesis
DD is a rare autosomal dominant disease affecting both sexes and all ethnic groups. Its prevalence has been estimated at 1 in 26,300 in Slovenia, 1 in 30,000 in Scotland,1 in 36,000 in northeast England, and 1 in 100,000 in Denmark. Penetrance of the disease is complete, and expression is highly variable between and within affected families. Spontaneous mutations are frequent.
The gene for DD was mapped by linkage analysis to chromosome band 12q23-24 in 1993, with no evidence for genetic heterogeneity in affected families. ATP2A2 was identified as the defective gene in 1999 using a candidate positional cloning approach. ATP2A2 encodes sarco/endoplasmic reticulum Ca2+ adenosine triphosphatase (ATPase) isoform 2 (SERCA2), a calcium pump transporting Ca2+ from the cytosol to the lumen of the ER. The identification of a Ca2+ pump as the defective protein in DD came as a surprise and shed light on the key role of Ca2+ signaling in the homeostasis of the epidermis.8
ATP2A2 spans 76 kilobases (kb), is organized in 21 exons, and encodes a 4.4-kb transcript, which is alternatively spliced into three isoforms: SERCA2a, SERCA2b, and SERCA2c. SERCA2a is expressed in the heart and slow-twitch skeletal muscles. SERCA2b and SERCA2c are ubiquitously expressed. SERCA2b is the major isoform detected in the human epidermis.11 SERCA2 pumps belong to the P-type Ca2+ ATPase family, defined by the highly conserved phosphorylation sequence DKTGT. They catalyze the hydrolysis of adenosine triphosphate (ATP) coupled with the translocation of 2 Ca2+ ions from the cytosol (100 nM) to the ER lumen, where Ca2+ is stored at high concentrations (500 µM).12 SERCA pumps comprise three cytoplasmic domains (the actuator, the phosphorylation, and the ATP-binding domains) linked by five stalk domains to 10 or 11 transmembrane domains anchored in the ER membrane. After binding of two Ca2+ ions, SERCA pumps undergo trans-phosphorylation from ATP, which leads to conformational changes and the release of Ca2+ ions into the ER lumen. This complex cycle involves critical interactions between cytoplasmic and transmembrane domains, four of which form the two Ca2+ ions binding pockets of the molecule. More than 130 ATP2A2 mutations have now been reported in DD patients, the majority of which are different from family to family (http://www.hgmd.cf.ac.uk/ac/gene.php?gene=ATP2A2). Mutations are distributed across the entire molecule. They disrupt critical functional domains of SERCA2 and show no evidence of clustering or mutation hot spots.
DARIER-WHITE DISEASE AT
AGLANCE
- Also called Darier disease or keratosis follicularis.
- Late-onset genetic disorder of keratinization.
- First described independently by Darier and White in 1889.
- Clinical features consist of warty papules and plaques in seborrheic areas, specific nail changes, palmo-plantar pits, and papules on the dorsum of the hands and feet.
- Histologic examination of skin lesions shows suprabasal acantholysis in the epidermis with dyskeratotic cells.
- Caused by loss-of-function mutations in the ATP2A2 gene encoding sarco/endoplasmic reticulum calcium adenosine triphosphatase isoform 2 (SERCA2), which impair intracellular Ca2+ signaling.
The majority of the mutations are missense mutations (50 percent) or inframe deletions or insertions (8 percent), which predict the synthesis of a structurally abnormal protein. Other mutations include nonsense mutations (12 percent) and frameshift mutations (23 percent), which lead to a premature termination codon (PTC) and predict loss of protein expression through nonsense messenger RNA (mRNA) decay. The remaining mutations are splice site mutations (7 percent), the effects of which on the mutated protein remain to be documented. Genotype-phenotype comparison initially suggested that ATP2A2 missense mutations are more frequent in severe or atypical forms, but this has not been confirmed in other studies.22 However, considerable inter- and intrafamilial variability in the disease phenotype indicates that additional factors influence disease expression.7,18,23 Three mutations identified in patients with classic DD are of specific interest because they occur in a region of exon 20 that is specific for SERCA2b isoform.13,28 Each of
these mutations leads to a PTC, which indicates that loss of SERCA2b expression is sufficient to cause DD and that SERCA2a isoform cannot compensate for it. This finding is consistent with the fact that the most prominent expression of SERCA2b is in the epidermis, although both isoforms are expressed in cultured keratinocytes.
The role of extracellular Ca2+ in keratinocyte adhesion and differentiation has been well documented, but the key function of SERCA2b in intracellular Ca2+ homeostasis in the epidermis was unsuspected. In normal keratinocytes, there is an increasing epidermal gradient in the normal skin, from the basal layer to the superficial layers. High extracellular concentrations are required for epidermal intercellular adhesion, differentiation, and cornification. Ca2+ signaling involves binding of raised extracellular Ca2+ to a Ca2+ plasma membrane receptor, which activates phospholipase C and generates inositol 1,4,5-tris-phosphate (IP3) and diacylglycerol from phosphatidylinositol bisphosphate (Fig. 49-1). IP3 in turn acts as a second messenger, binding to its receptors on the ER (and Golgi apparatus) to trigger release of Ca2+ from the intracellular stores into the cytoplasm. Emptying of ER Ca2+ stores activates an influx of extracellular Ca2+ through plasma membrane Ca2+ channels, which function like store-operated channels (capacitive entry mechanism).32 The molecular identity of these channels that amplify the Ca2+ response has not been clearly established, but they could include transient receptor potential channels.33 The SERCA2 pump plays a key role in Ca2+ signaling by refilling the internal Ca2+ reservoir of the ER, which is considered the major intracellular Ca2+ store. On stimulation, the release of Ca2+ from the
ER store will lead to specific cellular responses, depending on the amplitude, frequency, and sub-cellular location of the Ca2+ signal. Ca2+ triggers the switch between keratinocyte proliferation and differentiation. The increase of intracellular Ca2+ levels activates calmodulin, a major Ca2+-binding protein that plays a key role in the control of gene transcription by Ca2+. This results in the activation of the cytosolic calmodulin-dependent phosphatase calcineurin and the family of Ca2+/calmodulin-dependent protein kinases. Both types of enzymes contribute to the regulation of cell division and differentiation. Calcineurin dephosphorylates the cytoplasmic NFAT (nuclear factor of activated T cells) proteins, which allows their translocation into the nucleus and the induction of their downstream target genes. It is of interest to note that cyclosporin A, tacrolimus, and pimecrolimus are calcineurin inhibitors (see Chap. 221). Activation of the Ca2+/calmodulin-dependent protein kinase cascade involves three distinct kinases that regulate transcription through phosphorylation of nuclear factors and histone acetylation, which leads to transcriptional activation or inhibition. By keeping appropriate Ca2+ concentrations in the ER lumen, SERCA2 also plays an essential role in protein synthesis, chaperone-dependent processing, and post-translational modification of membrane and secreted proteins, which require a unique calcium-rich environment. Ca2+ is required for the assembly of desmosomes and adherens junctions as well as actin polymerization.
A change in the Ca2+ ER luminal content is likely to have a major effect on Ca2+ signaling, protein post-translational modifications, and trafficking. Although no glycosylation defect of the desmosomal proteins was detected in DD keratinocytes, in culture trafficking of desmoplakin to the membrane is significantly hindered.42 Defective expression of this key molecule that links the cytoskeleton to the desmosomal complexes could contribute to impaired cell-to-cell adhesion in the epidermis in DD. Finally, Ca2+ plays a key role in the transcriptional regulation of a wide range of genes that are essential for epidermal cell-to-cell adhesion and terminal differentiation, which suggests that abnormal Ca2+ signaling could have a profound impact on the transcription of these genes.
The observation that exposure to ultraviolet B (UVB) irradiation, heat, and infection triggers disease manifestations indicates the role of external factors in unmasking the basic defect in DD. In the absence of stress, SERCA2 deficiency can be compensated by increased expression of the normal allele and/or other regulatory systems. Triggering factors would disrupt this subtle balance by downregulating ATP2A2 or increasing the requirement for SERCA2 to maintain a unique Ca2+ content in the lumen. Because only one copy of ATP2A2 is functional, reduction of SERCA2 levels would be excessive, or compensation would not reach adequate levels. Consistent with this possibility, UVB irradiation and proinflammatory cytokines have been shown to downregulate ATP2A2 as well as ATP2C1 mRNA expression.43 Reduced levels of SERCA2 would result in inappropriate Ca2+ concentrations in the lumen, causing dysfunction of calcium-dependent chaperone molecules that leads to impair folding, assembly, and/or trafficking of proteins. Whether this preferentially affects proteins playing a key role in cell-to-cell adhesion and/or differentiation or affects protein processing in general remains to be determined.
Clinical Findings
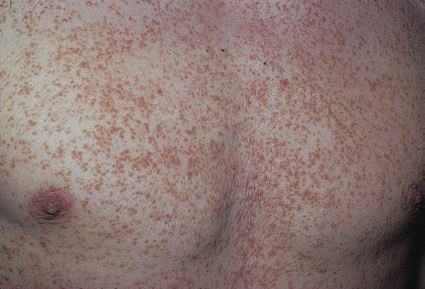
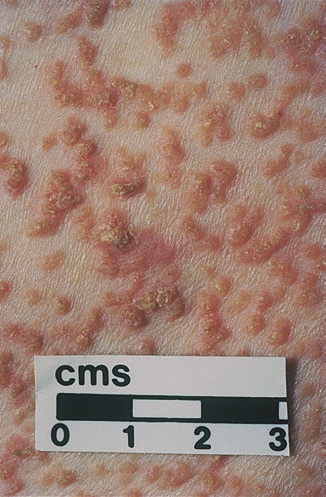
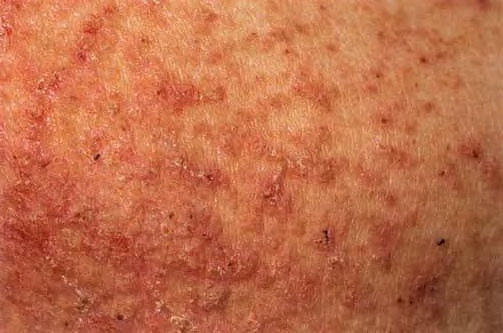
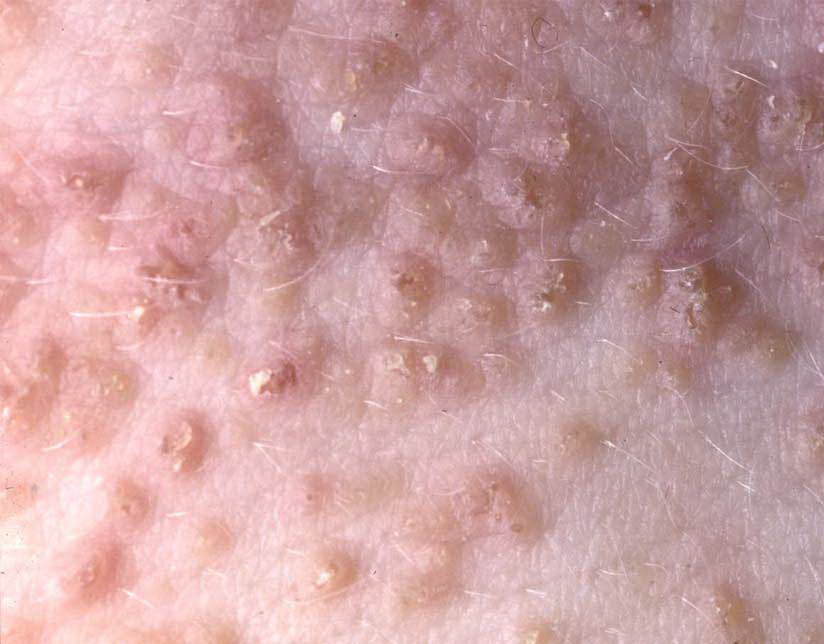
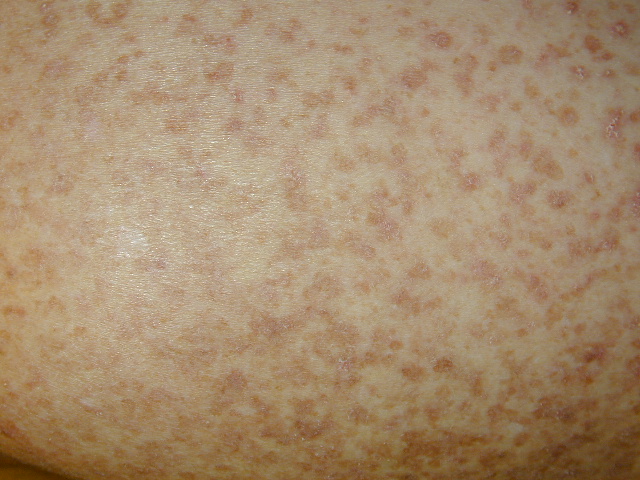
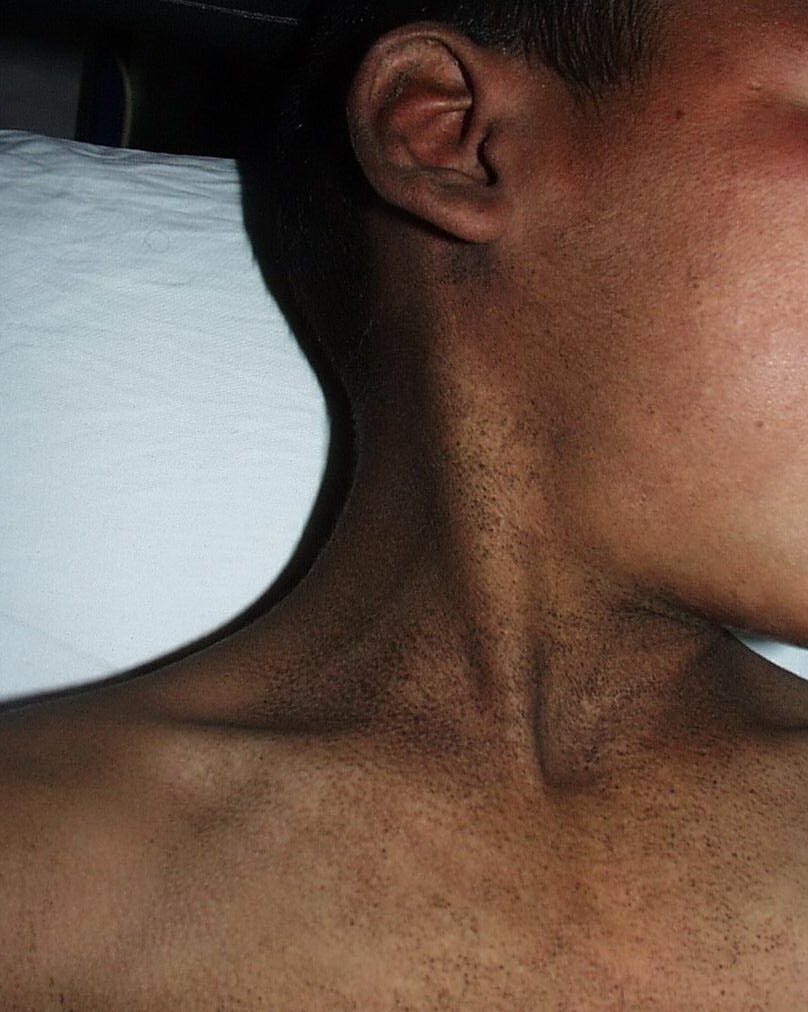
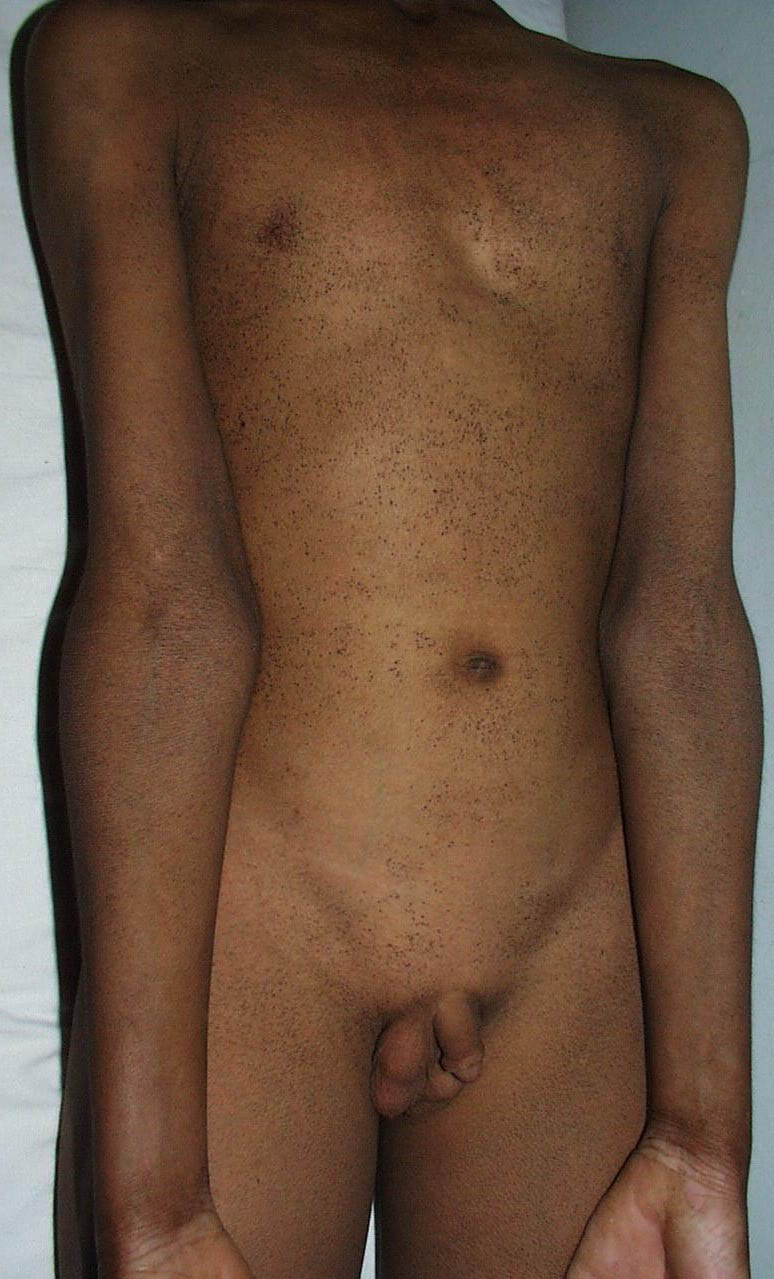
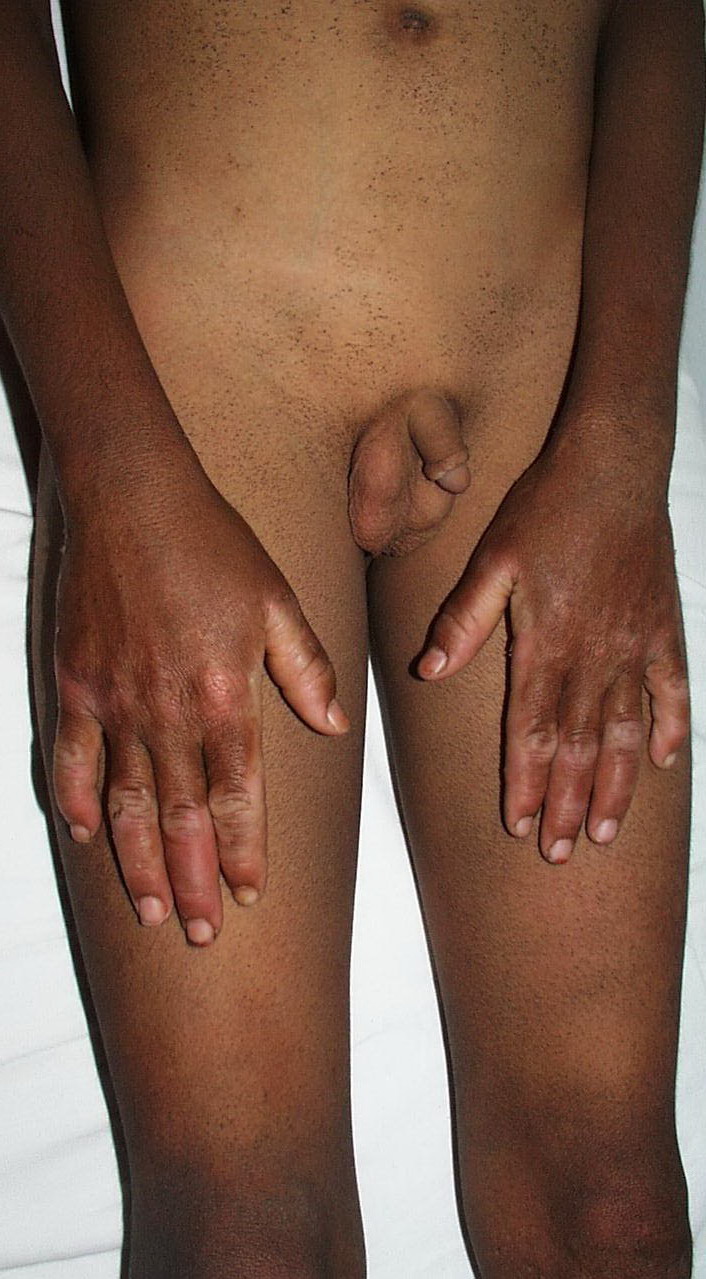
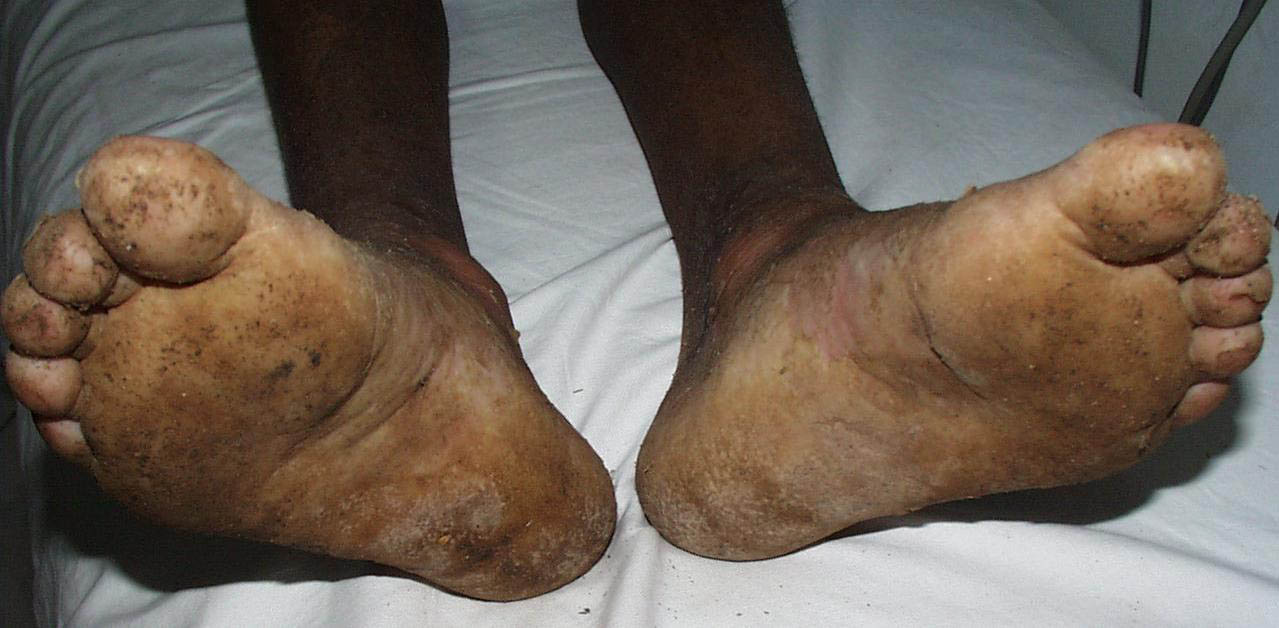
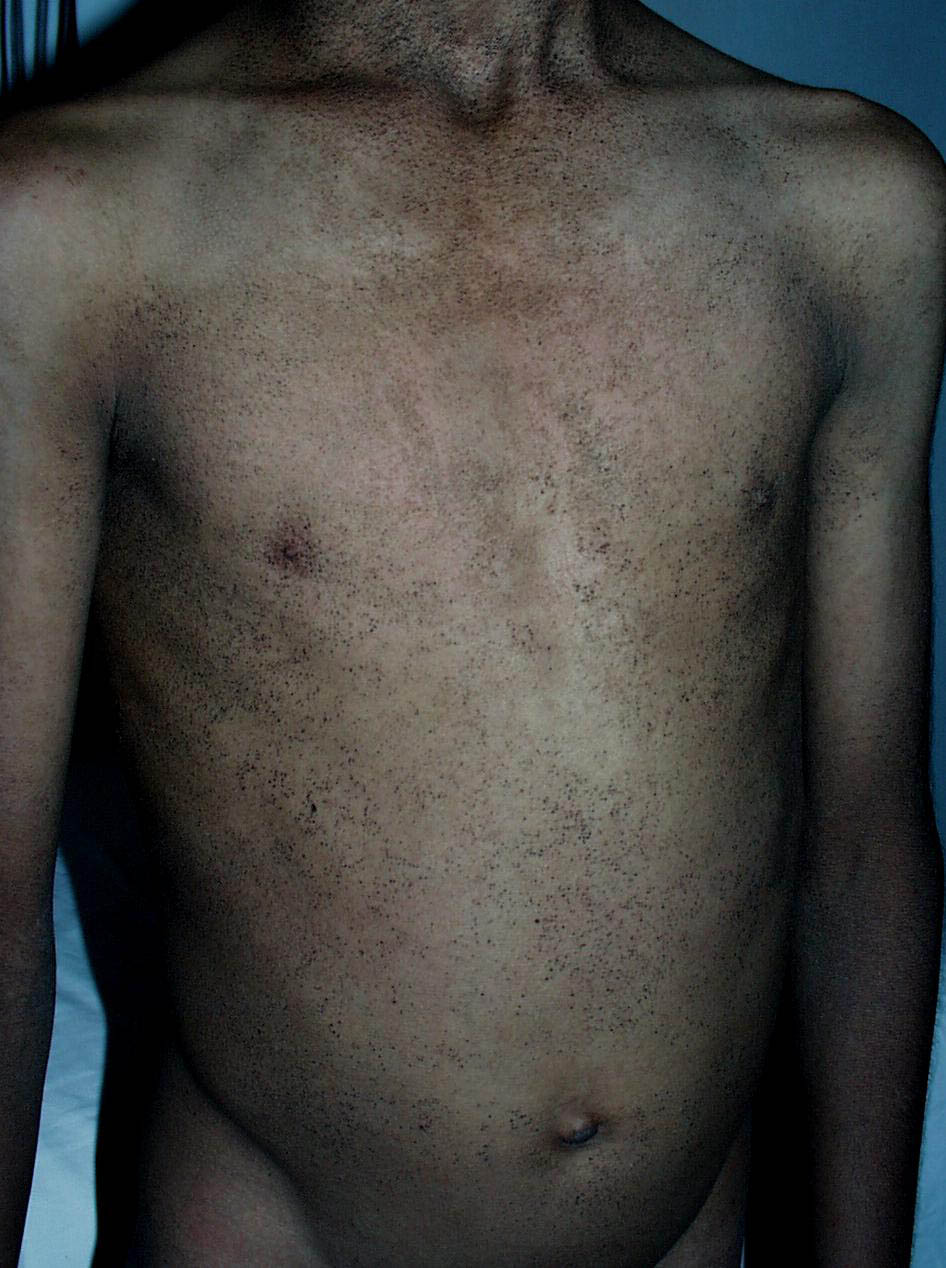
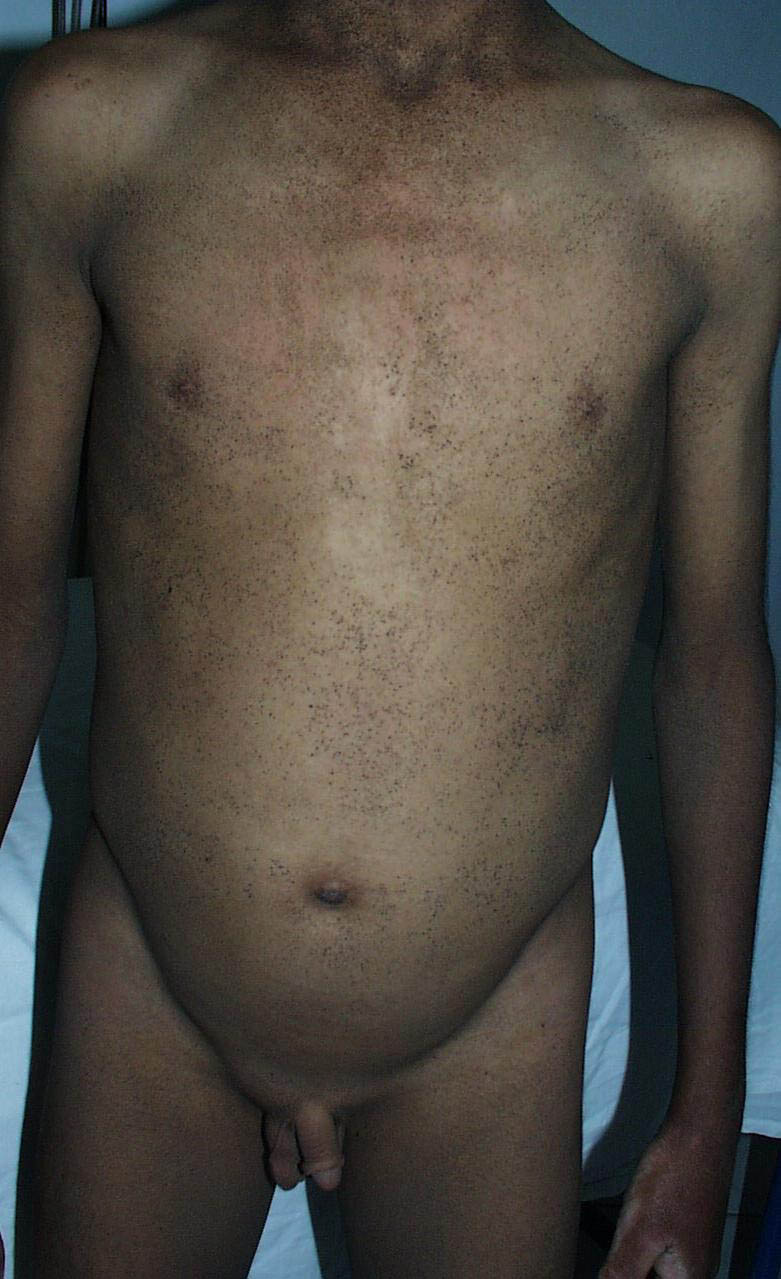
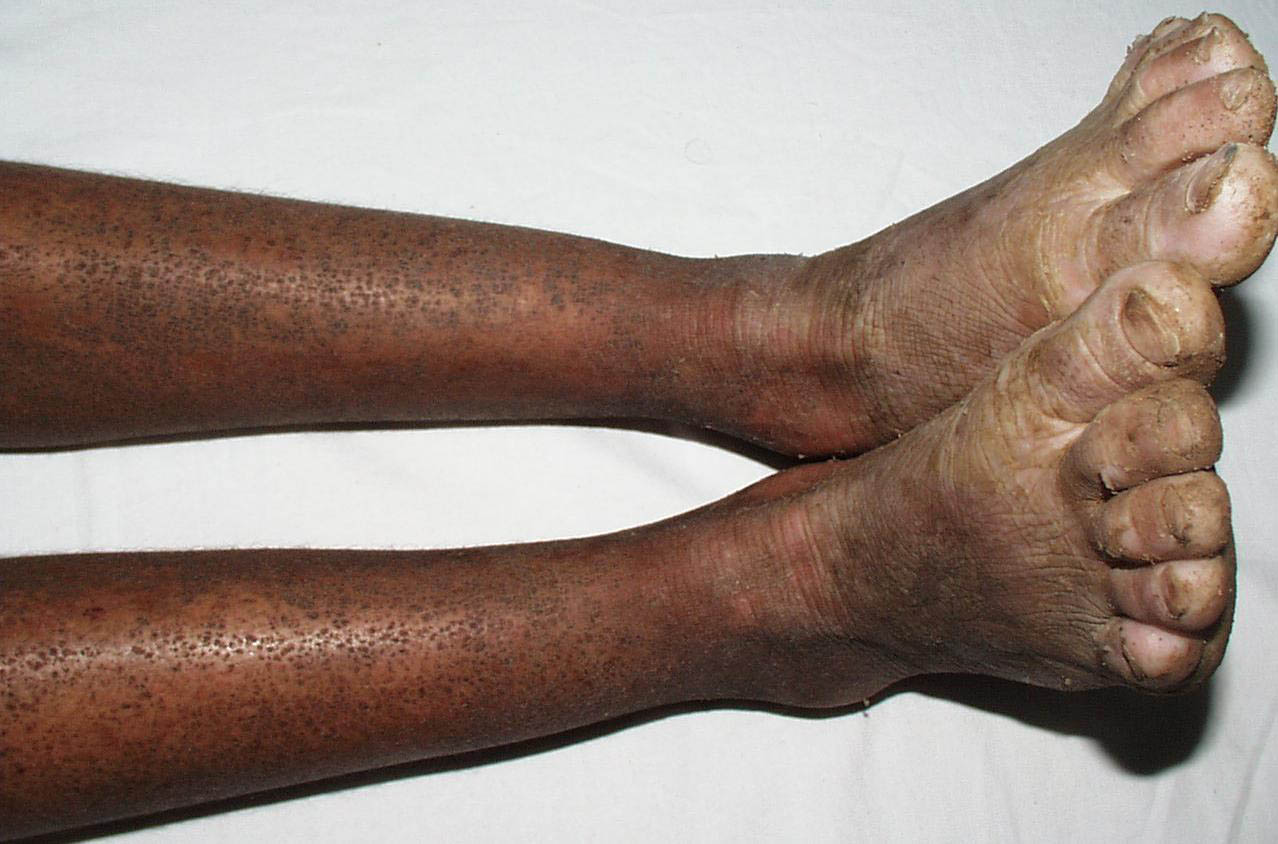
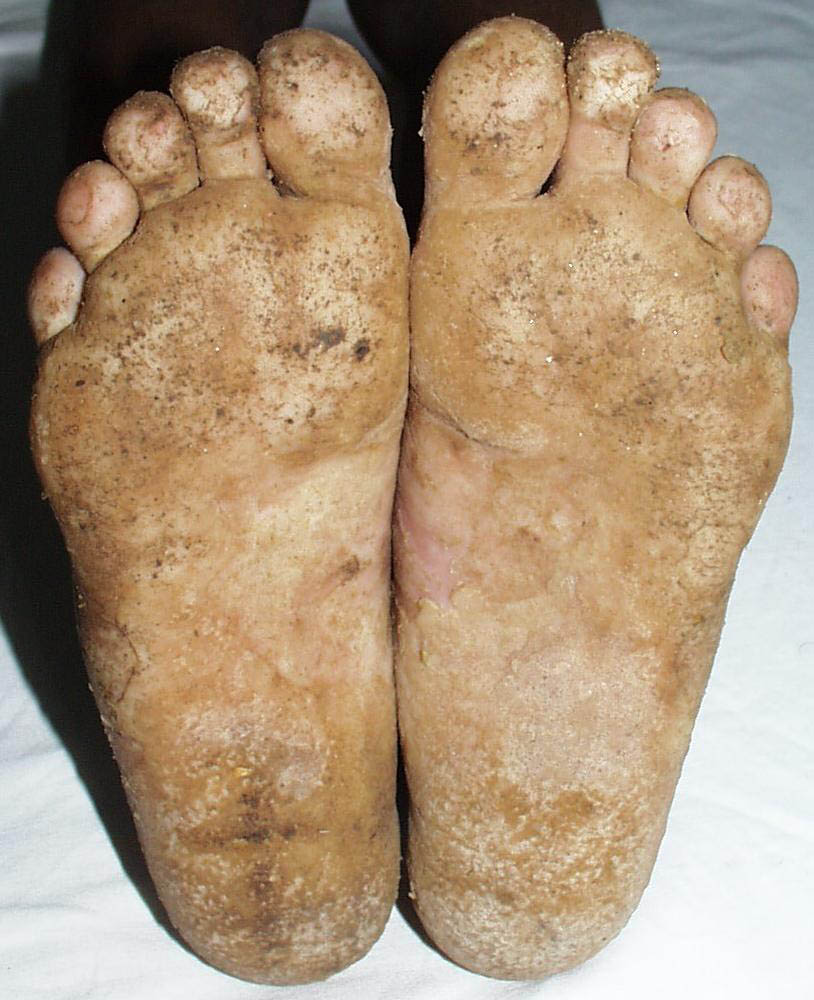
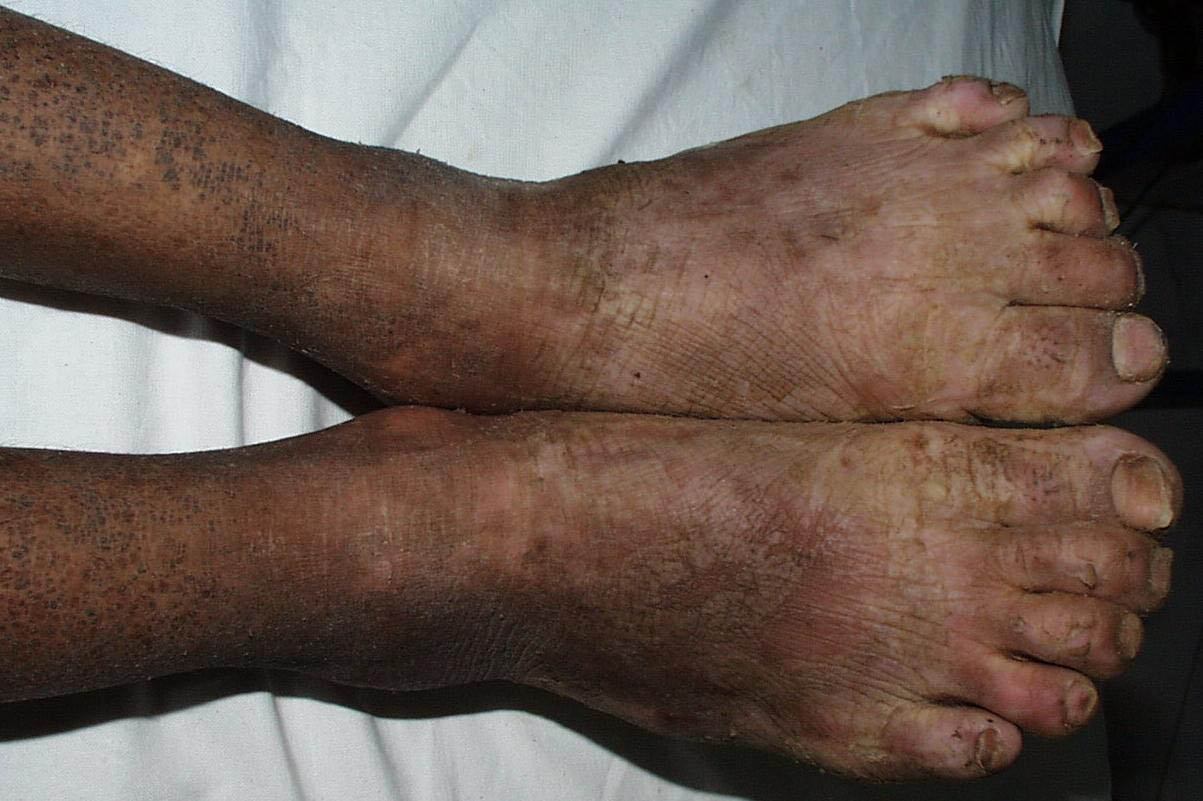
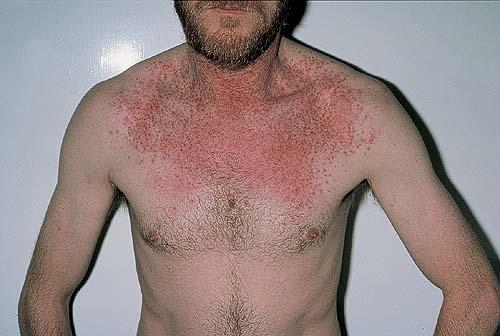
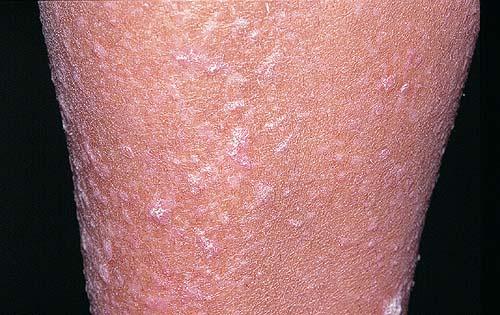
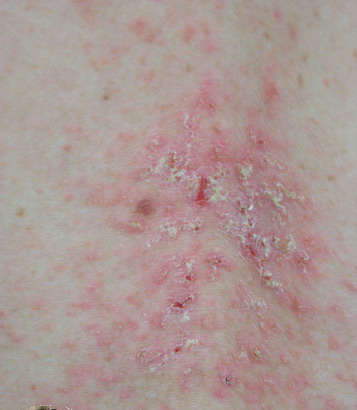
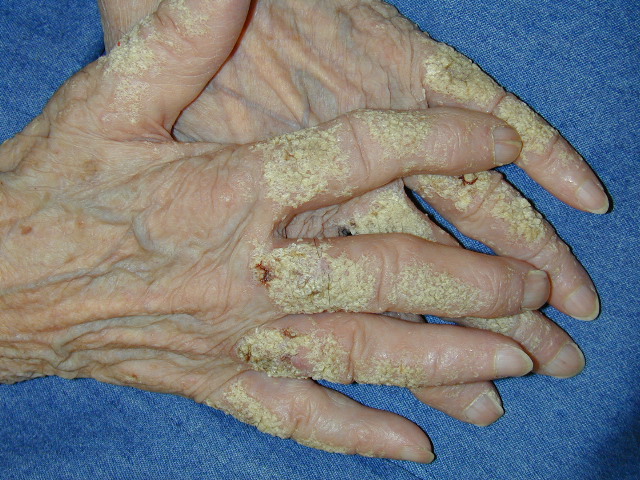
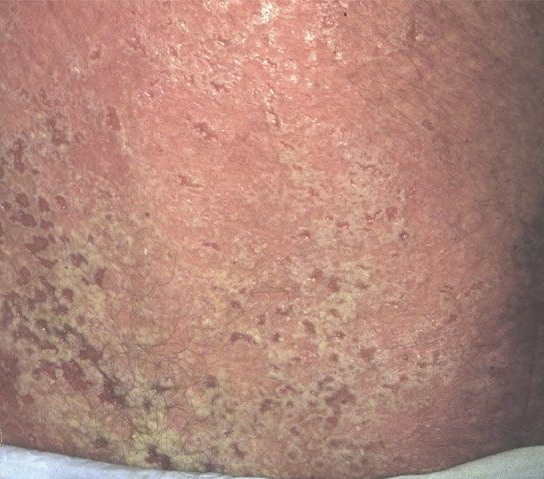
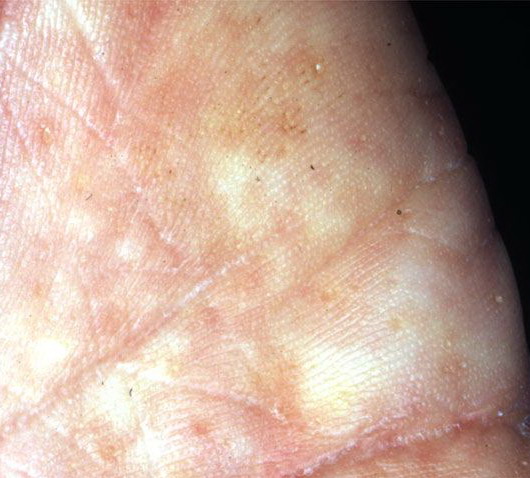
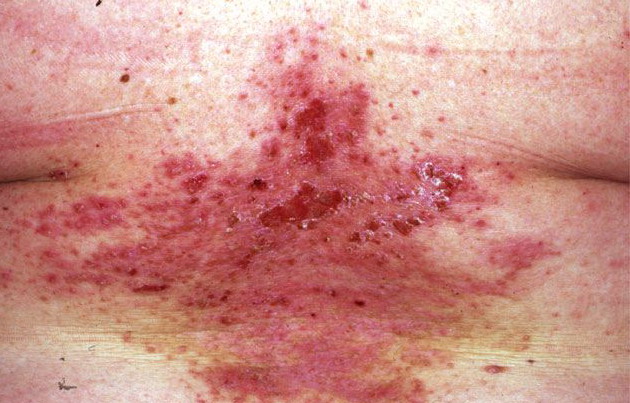
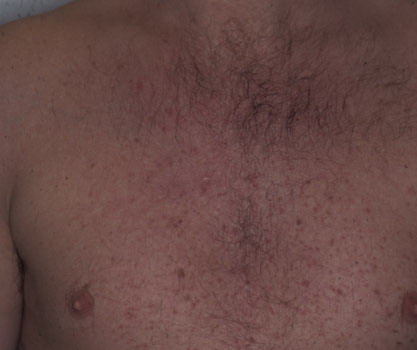
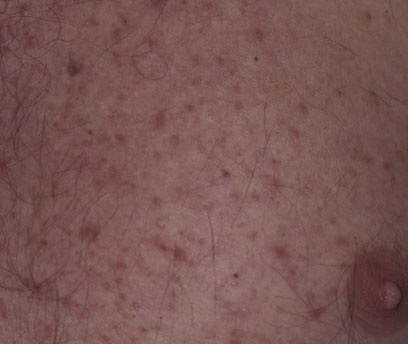
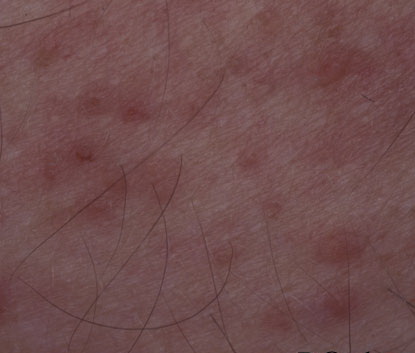
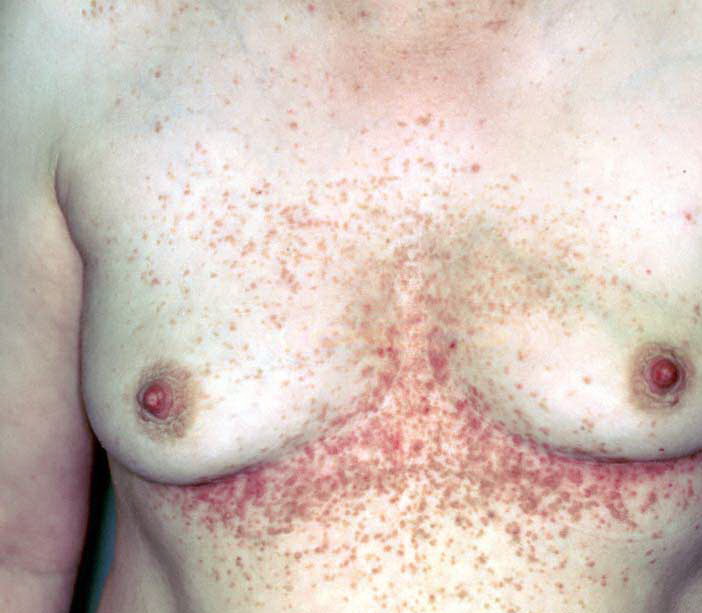
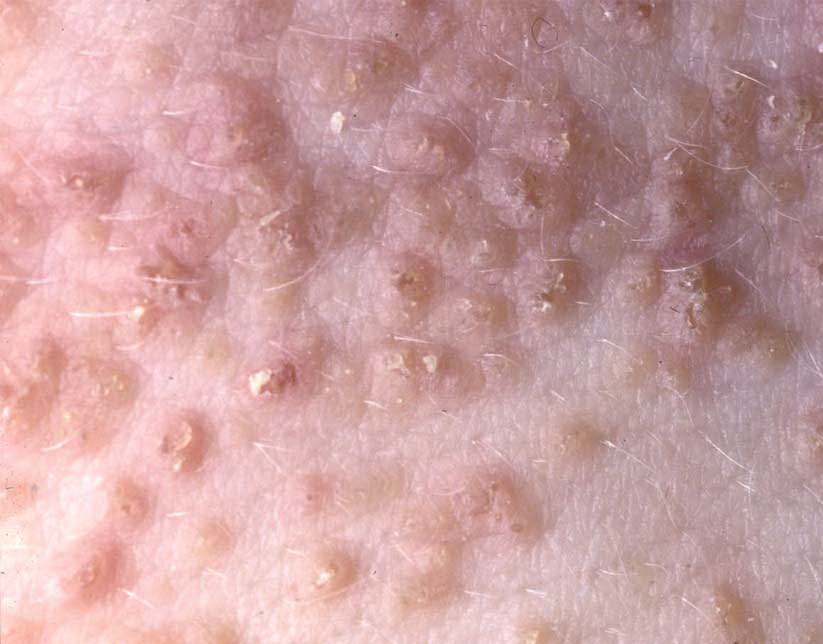
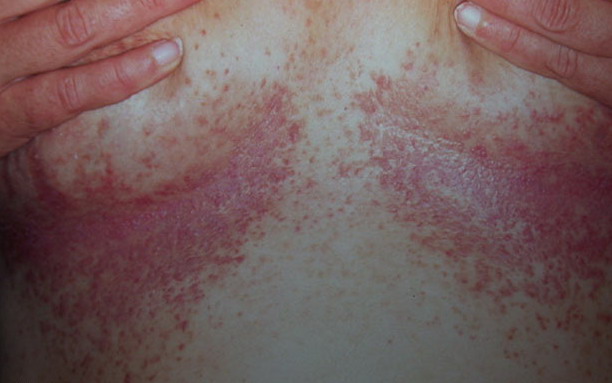
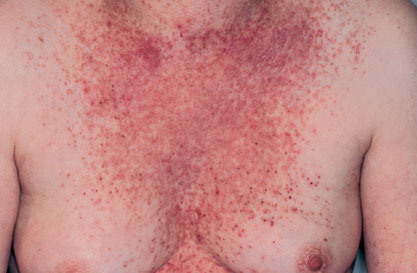
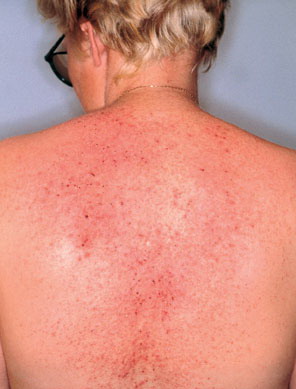
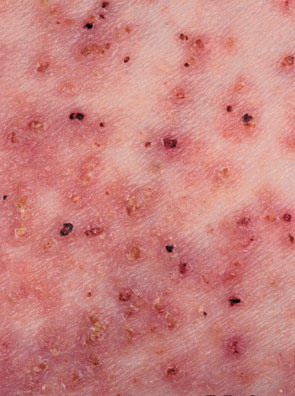
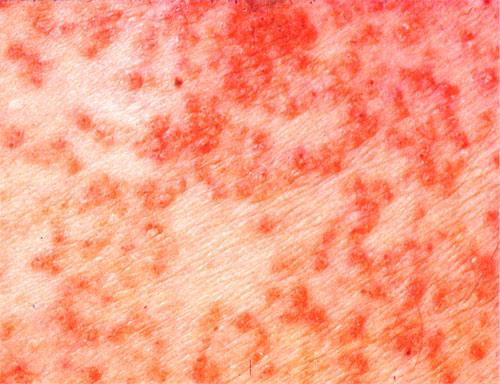
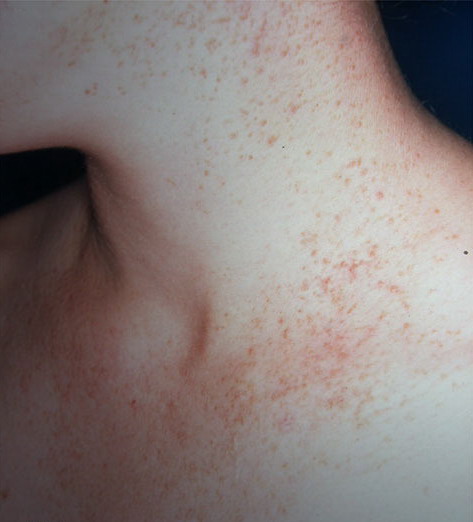
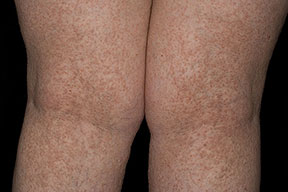
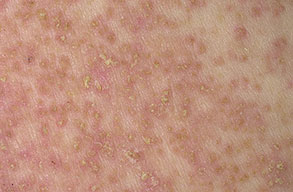
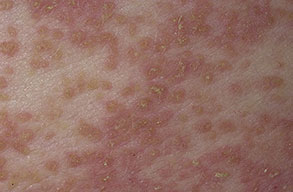
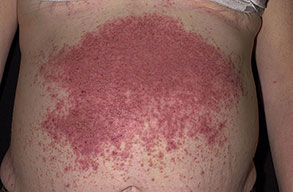
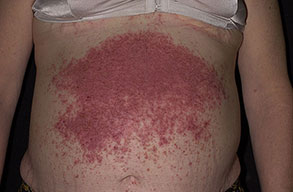
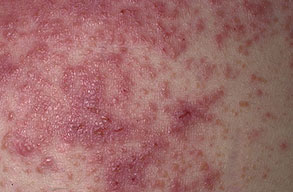
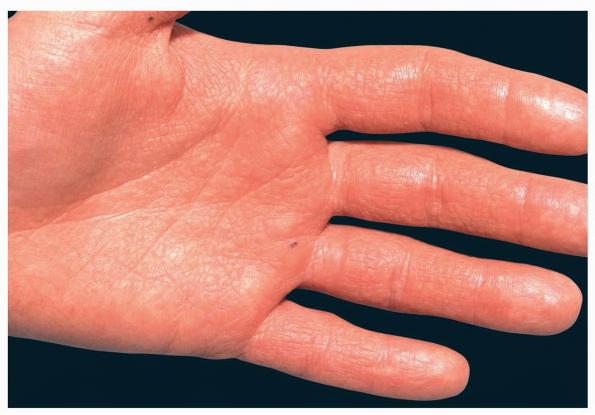
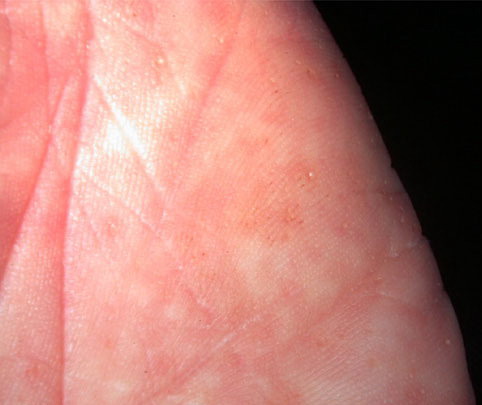
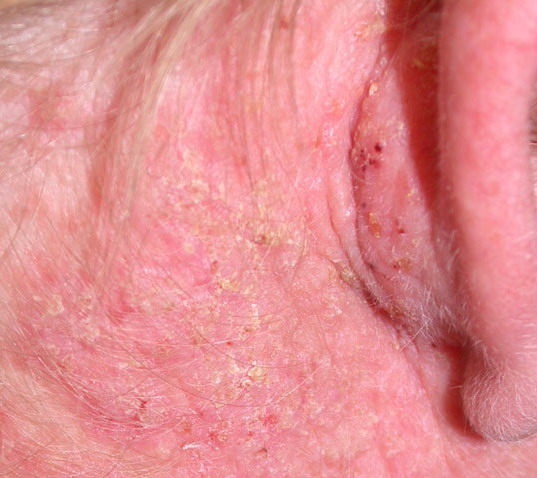
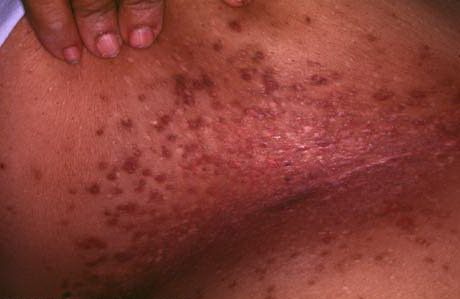
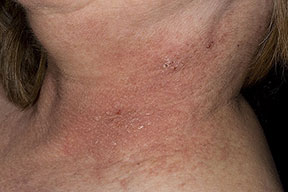
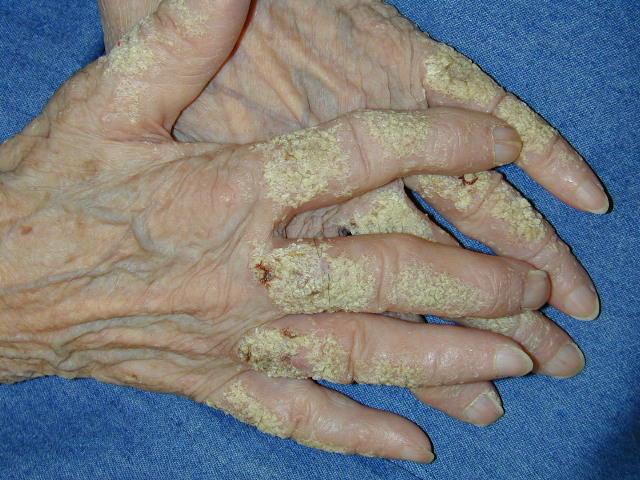
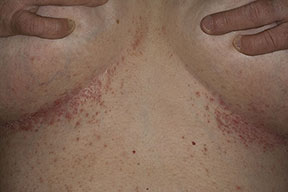
Onset of the disease is usually around puberty. The majority of patients develop the first lesions between 10 and 20 years of age. Typical lesions are greasy keratotic papules, skin-colored, yellow-brown or brown, which may be isolated or may form relatively large, crusted, and confluent plaques. The sites of predilection are the seborrheic areas of the trunk and face: the upper chest, the back, the sides of the neck, the forehead, the ears, and the scalp. The groin, axillae , and anogenital region are also frequently involved. The skin lesions can become vegetating in the folds and are often infected, malodorous, and responsible for major discomfort. They can be limited or form extensive plaques during acute phases. Careful examination of the palms and soles will frequently reveal small pits or punctuated keratoses, which are highly suggestive of DD . Hands and feet can also show discrete flat, skin-colored papules on the dorsal areas, similar to AKV of Hopf.4
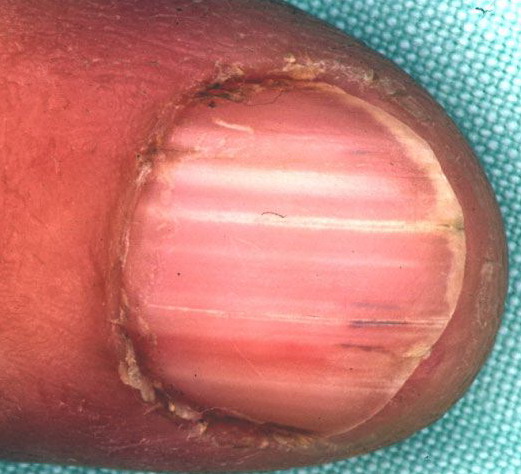
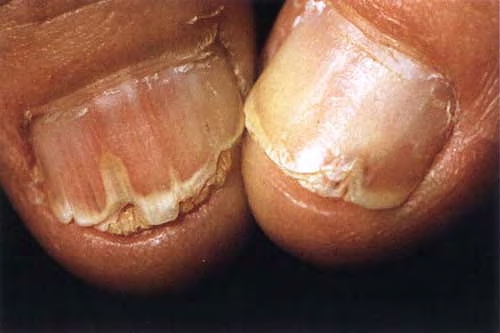
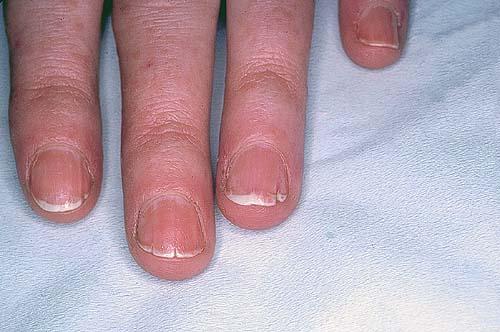
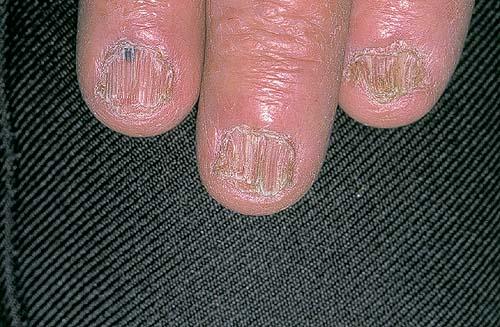
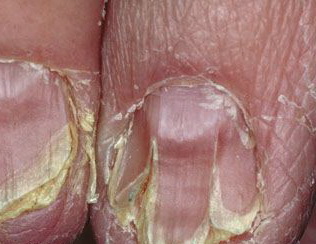
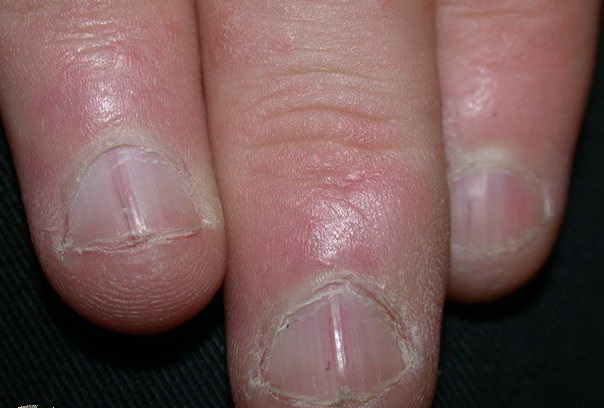
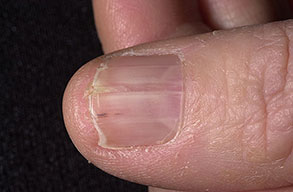
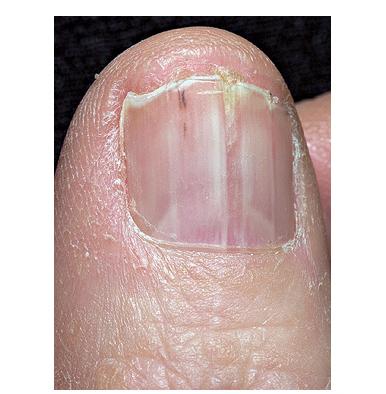
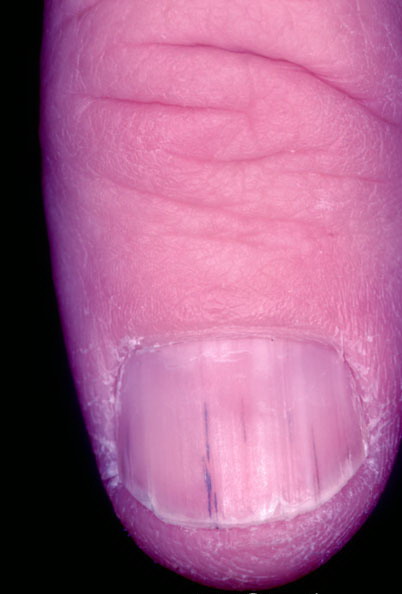
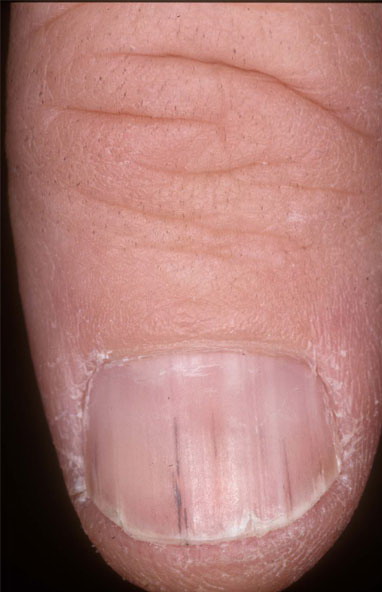
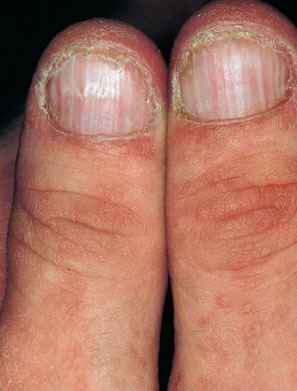
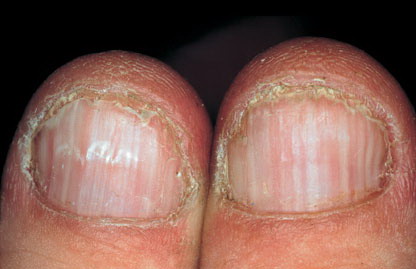
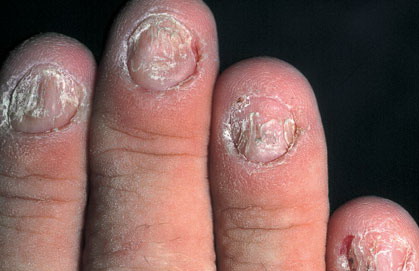
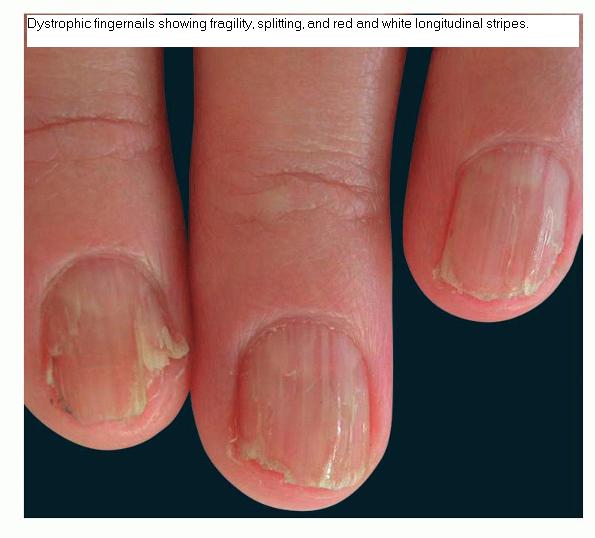
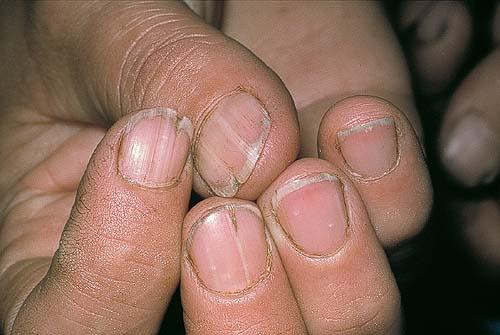
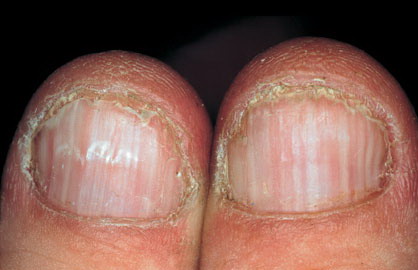
Nail abnormalities are an almost constant feature and are highly specific for
the disease. The nails are fragile and split easily. They show a combination of red and white longitudinal stripes, have a V-shaped nick at the free margin of the nail, and show subungual hyperkeratosis .
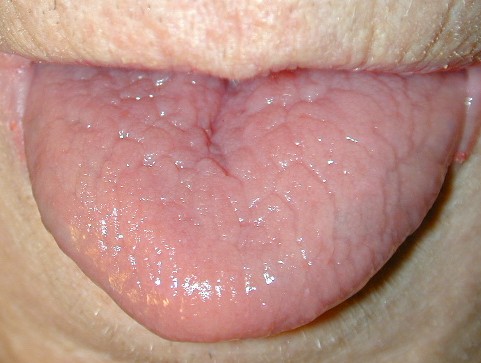
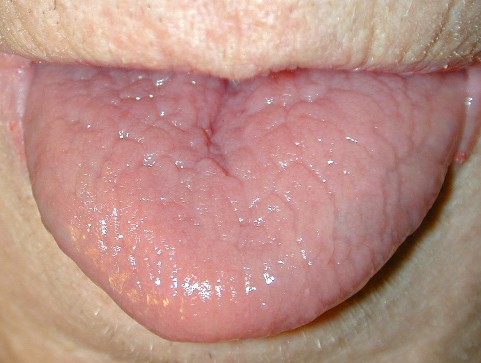
The mucous membranes also can be affected. The hard palate, oral mucosa, gingivae, esophagus, vulva, and rectum may be sites of whitish small papules, often densely grouped (leukoplakia).44 Blockage of the salivary glands has been reported. Intrafamilial differences in disease severity have been described in several studies, which indicates that additional genetic and/or environmental factors influence disease expression.
Histopathology
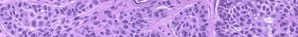
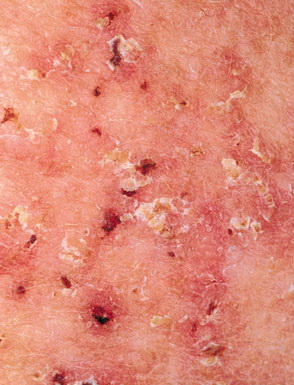
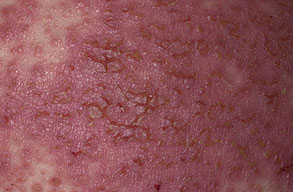
Histologic examination shows hyperkeratosis, focal dyskeratosis (premature and abnormal keratinization of single keratinocytes) associated with suprabasal acantholysis leading to suprabasal clefts . The rounded eosinophilic dyskeratotic cells are called corps ronds in the stratum spinosum, and grains in the upper layers of the epidermis. They are thought to correspond to apoptotic keratinocytes. Electron microscopy has revealed separation of the keratin filaments from the desmosomes with clumping of perinuclear tonofilaments.
Associated Manifestations
Behavior and learning difficulties have been observed in patients with DD, but they may be, at least in part, secondary to the social handicap caused by the disease. Neuro-psychiatric abnormalities have also been reported in families with DD. These include seizures, mild mental retardation, bipolar disease, and schizophrenia. Familial co-segregation of DD and bipolar disorder has been reported in several families and strongly supports the existence of a bipolar disorder susceptibility gene in the DD region. However, ATP2A2 has been excluded as a common susceptibility gene for bipolar disease. These observations, together with the finding of SERCA2b expression in the brain49 and the importance of calcium signaling in neurons, raise the question of whether skin and neuro-psychiatric conditions are occurring independently or represent expression of the same genetic defect affecting both skin and brain.
Clinical Forms
Some patients develop erosive or bullous forms; others have malodorous, vegetating lesions in the flexures. Pruritic and papulovesicular lesions can be seen in some DD patients, similar to those in GD (see Grover Disease). Rare clinical variants include cornifying forms on the legs, comedone-like lesions, and hemorrhagic macules on palms and soles. In pigmented skin, DD may present as hypopigmented macules and papules.
Two particular forms of the disease should be distinguished. In the first,
acantholytic dyskeratotic epidermal nevi, the localized and unilateral keratotic papules of DD follow Blaschko's lines . Molecular analysis in these patients has shown that they represent mosaic forms of DD, carrying a postzygotic ATP2A2 mutation in affected areas only. There has been no report of patients with segmental DD having a child with generalized DD, and the risk of transmission of a generalized form remains unknown.
Acral forms are difficult to distinguish from AKV of Hopf, which could be considered a clinical variant of DD, because both diseases are due to ATP2A2 mutations (see Acrokeratosis Verruciformis).
Differential Diagnosis
The classic form of DD is highly suggestive of the condition. However, DD can be misdiagnosed as seborrheic dermatitis or acne in the initial stages. The diagnosis of clinical variants can sometimes be challenging . Acral forms resemble plane warts. Erosive or bullous forms predominating in the folds may be difficult to distinguish from HHD (see Hailey-Hailey Disease) and pemphigus vulgaris . However, HHD usually presents later in the third decade and shows painful flexural erosions with rhagades and no keratotic papules. Vegetating forms need to be distinguished from pemphigus vegetans . Pruritic and papulovesicular forms resemble GD. However, GD occurs in patients with long-term sun damage and is usually transient, although chronic forms have been described.
Course
The skin lesions generally appear between early childhood and the age of 20 years. The condition runs a chronic relapsing course, with exacerbations throughout life. Some patients will have relatively mild disease, whereas others, sometimes within the same family, will develop a more severe form. In particular, the skin lesions are exacerbated by exposure to sunlight or artificial UVB radiation, heat, sweating, friction, and infections. Patients with DD appear to have an increased susceptibility to herpes simplex and chronic pyogenic infections. Rare cases of recurrent corneal ulcerations with perforation have been reported.
Treatment
Patients with mild disease will benefit from advice about sun protection (sun avoidance and use of sunscreens in the summer) and avoidance of heat. When skin lesions are limited, emollients containing urea or lactic acid can reduce crusting. Topical applications of tretinoin or isotretinoin improve the hyperkeratosis of localized lesions, although irritation limits their value. Topical steroids may help in reducing irritation but are not effective alone. New retinoids such as tazarotene are somewhat effective and are tolerated better than first-generation retinoids. Antiseptics (topical or in the bath), antibiotics, and antifungal medications are helpful to prevent or treat pyogenic and fungal infection. Herpes infection should be suspected when lesions are painful and exacerbated and requires antiviral treatment with acyclovir.
Box 49-1 Differential Diagnosis of Darier Disease
- Classic form
- Seborrheic dermatitis
- Seborrheic keratoses
- Candida infection (submammary)
- Papular vesicular form
- Erosive, bullous form
- Hailey-Hailey disease
- Pemphigus vulgaris
- Langerhans cell histiocytosis
- Candida infection
- Impetigo
- Vegetating form
- Acral form
For those with more severe disease, oral retinoids such as acitretin are the most effective treatment. Their efficacy lasts only as long as the treatment is continued. The initial dose is usually 25 to 30 mg per day for 2 to 4 weeks, and if the drug is well tolerated, the dose can be increased up to 60 mg for 2 months. The patient must be monitored carefully for possible side effects . After initial clearing of the lesions, the dosage should be reduced progressively and stopped (in winter) or maintained at as low a level as possible to avoid relapse.4 Topical tacrolimus has been used successfully to treat extensive DD.
The disease can cause a considerable social handicap and can require specific psychological support by a medical psychologist. Genetic counseling should be offered, although prenatal diagnosis of the disease should be restricted to extremely disabling forms.