Cornelia de Lange syndrome is a developmental disorder that affects many parts of the body. The features of this disorder vary widely among affected individuals and range from relatively mild to severe.
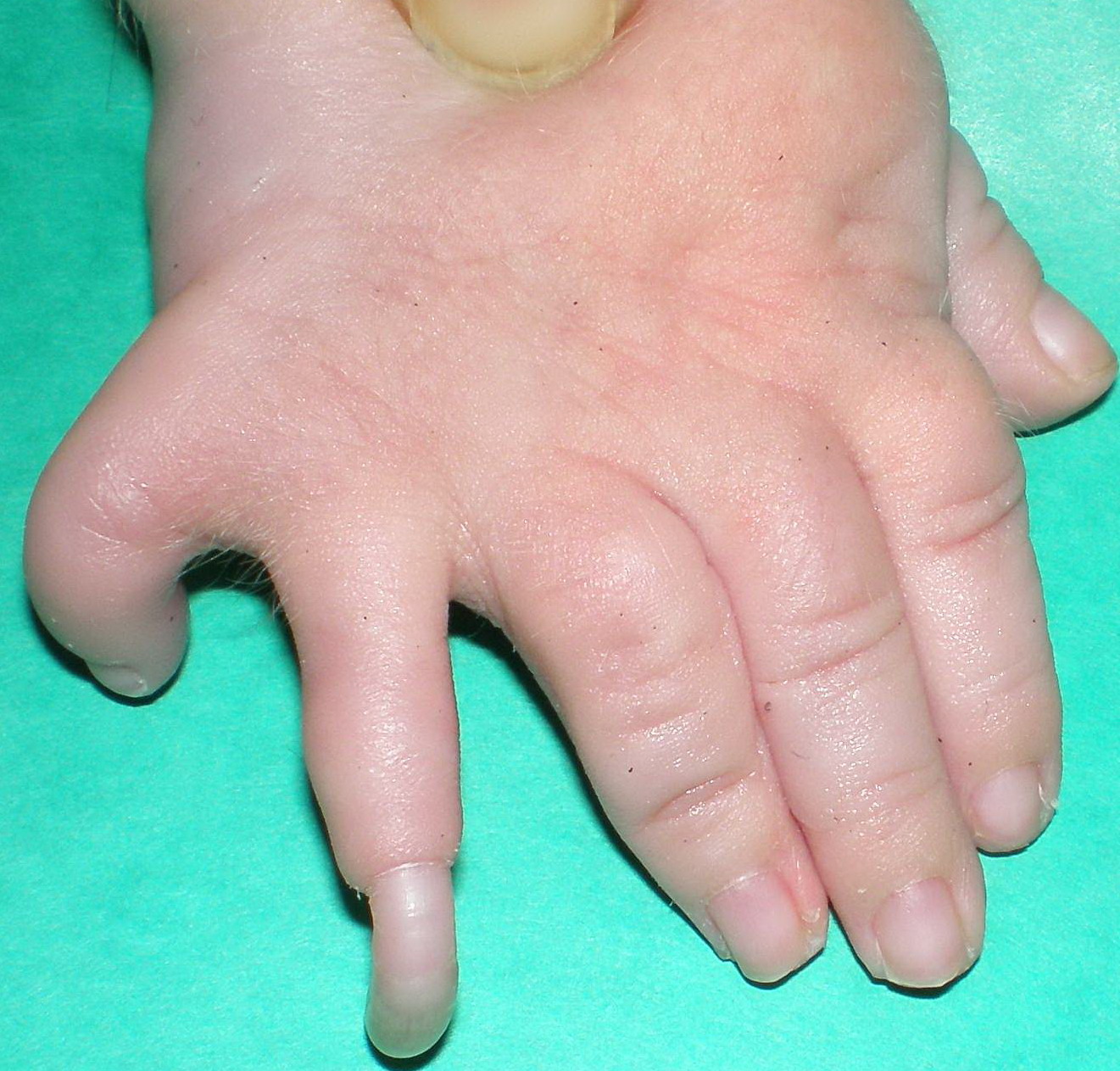
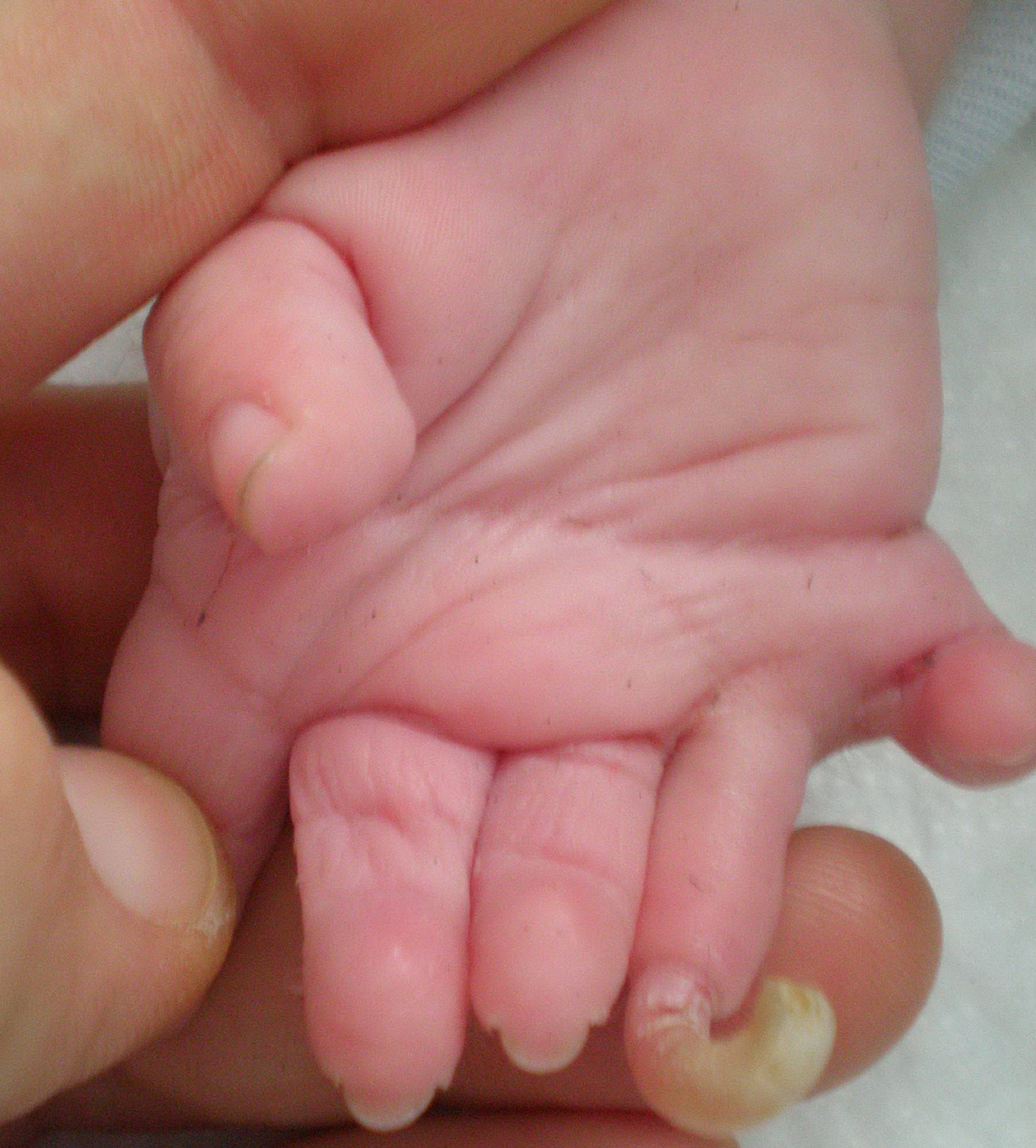
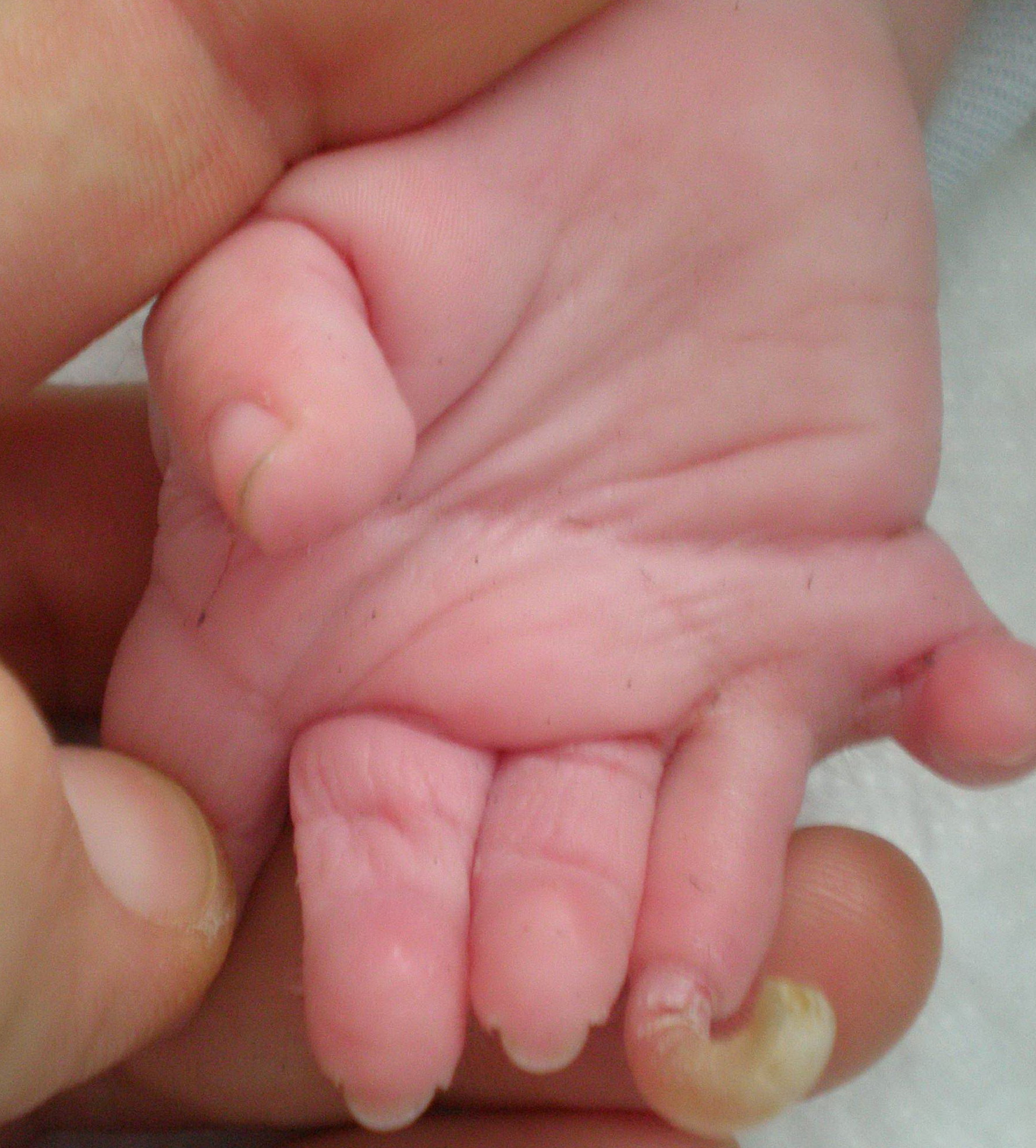
Cornelia de Lange syndrome is characterized by slow growth before and after birth, intellectual disability that is usually severe to profound, skeletal abnormalities involving the arms and hands, and distinctive facial features. The facial differences include arched eyebrows that often grow together in the middle (synophrys); long eyelashes; low-set ears; small, widely spaced teeth; and a small, upturned nose. Many affected individuals also have behavior problems similar to autism, a developmental condition that affects communication and social interaction.
Additional signs and symptoms of Cornelia de Lange syndrome can include excessive body hair (hirsutism), an unusually small head (microcephaly), hearing loss, short stature, and problems with the digestive tract. Some people with this condition are born with an opening in the roof of the mouth called a cleft palate. Seizures, heart defects, eye problems, and skeletal abnormalities also have been reported in people with this condition.
Although the exact incidence is unknown, Cornelia de Lange syndrome likely affects 1 in 10,000 to 30,000 newborns.
Mutations in the NIPBL, SMC1A, and SMC3 genes can cause Cornelia de Lange syndrome. NIPBL gene mutations have been identified in more than half of all people with this condition; mutations in the other two genes are much less common. The proteins produced from all three genes play important roles in directing development before birth. Within cells, these proteins help regulate the structure and organization of chromosomes and are involved in the repair of damaged DNA. They also regulate the activity of certain genes in the developing limbs, face, and other parts of the body.
Mutations in the NIPBL, SMC1A, and SMC3 genes can cause Cornelia de Lange syndrome by disrupting gene regulation during critical stages of early development. Studies suggest that SMC1A and SMC3 gene mutations tend to cause somewhat milder signs and symptoms than those seen with mutations in the NIPBL gene.
In about 35 percent of cases, the cause of Cornelia de Lange syndrome is unknown. Researchers are looking for additional changes in the NIPBL, SMC1A, and SMC3 genes, as well as mutations in other genes, that may be responsible for this condition.
When Cornelia de Lange syndrome is caused by mutations in the NIPBL or SMC3 gene, this condition is considered to have an autosomal dominant pattern of inheritance. Autosomal dominant inheritance means one copy of the altered gene in each cell is sufficient to cause the disorder. Almost all cases result from new gene mutations and occur in people with no history of the condition in their family.
Cases of Cornelia de Lange syndrome caused by SMC1A gene mutations have an X-linked pattern of inheritance. A condition is considered X-linked if the mutated gene that causes the disorder is located on the X chromosome, one of the two sex chromosomes. Studies of X-linked Cornelia de Lange syndrome indicate that one copy of the altered gene in each cell may be sufficient to cause the condition. Unlike most X-linked conditions, in which males are more frequently affected or experience more severe symptoms than females, X-linked Cornelia de Lange syndrome appears to affect males and females similarly. Most cases result from new mutations in the SMC1A gene and occur in people with no history of the condition in their family.
.