PITYRIASIS LICHENOIDES
Epidemiology
Pityriasis lichenoides affects all racial and ethnic groups in all geographic regions. It is more common in children and young adults but can affect all ages. PLC is three to six times more common than PLEVA.
Etiology and Pathogenesis
The etiology of pityriasis lichenoides is unknown. Some cases have been associated with infectious agents such as Toxoplasma gondii, Epstein-Barr virus, cytomegalovirus, parvovirus B19, and human immunodeficiency virus. At least one case was linked repeatedly with estrogen-progesterone therapy and another with chemotherapy drugs. It is uncertain whether these agents are actively involved in disease pathogenesis or merely coincidental bystanders; however, several cases associated with toxoplasmosis have cleared fairly quickly in response to specific therapy.
Immunohistologic studies have shown a reduction in CD1a+ antigen-presenting dendritic (Langerhans) cells within the central epidermis of pityriasis lichenoides lesions.69 Keratinocytes and endothelial cells are HLA-DR+, which suggests activation by T-cell cytokines. CD8+ T cells predominate in PLEVA, whereas either CD8+ or CD4+ T cells predominate in PLC. Many of these T cells express memory proteins (CD45RO) and cytolytic proteins (TIA-1 and granzyme B). Dominant T-cell clonality has been demonstrated in about half of PLEVA cases and a minority of PLC cases. In aggregate, these findings raise the possibility that pityriasis lichenoides is a variably clonal cytolytic memory T-cell lymphoproliferative response to one or more foreign antigens. Deposition of immunoglobulin M, C3, and fibrin in and around blood vessels and along the dermoepidermal
junction in early acute lesions suggests a possible concomitant humoral immune response, although this could be a secondary phenomenon.
The relationship of pityriasis lichenoides to lymphomatoid papulosis remains controversial. Common features include dominant T-cell clonality and spontaneous resolution of papular, predominantly lymphoid lesions. Furthermore, individual lesions with the clinicopathologic characteristics of either pityriasis lichenoides or lymphomatoid papulosis can co-exist in the same patient, either concurrently or serially. It remains to be determined whether this can be explained as an artifact of sampling lymphomatoid papulosis lesions at various stages of their evolution. The presence of large CD30+ atypical lymphoid cells is the hallmark of lymphomatoid papulosis (at least types A and C).74 Furthermore, these cells are typically CD4+ and often lack one or more mature T-cell antigens such as CD2, CD3, and CD5. These features serve to distinguish lymphomatoid papulosis from pityriasis lichenoides. Although occasional CD30+ cells can be seen in a wide variety of dermatoses, the presence of any appreciable number should favor lymphomatoid papulosis over pityriasis lichenoides as a matter of definition. It may be that the “PLCPLEVA” and “lymphomatoid papulosis-CD30+ anaplastic large cell lymphoma” disease spectra are intersecting rather than overlapping entities; that is, although pityriasis lichenoides is a distinct cutaneous T-cell disorder, it is possible that it may sometimes serve as fertile soil for the development of the CD30+ T-cell clone characteristic of lymphomatoid papulosis.
Clinical Findings
CUTANEOUS LESIONS
PLC and PLEVA exist on a clinicopathologic continuum. Therefore, individual patients may exhibit a mixture of acute and chronic lesions sequentially or concurrently. In addition, lesions representing clinical or histopathologic intergrades between the extremes may also occur at any time.
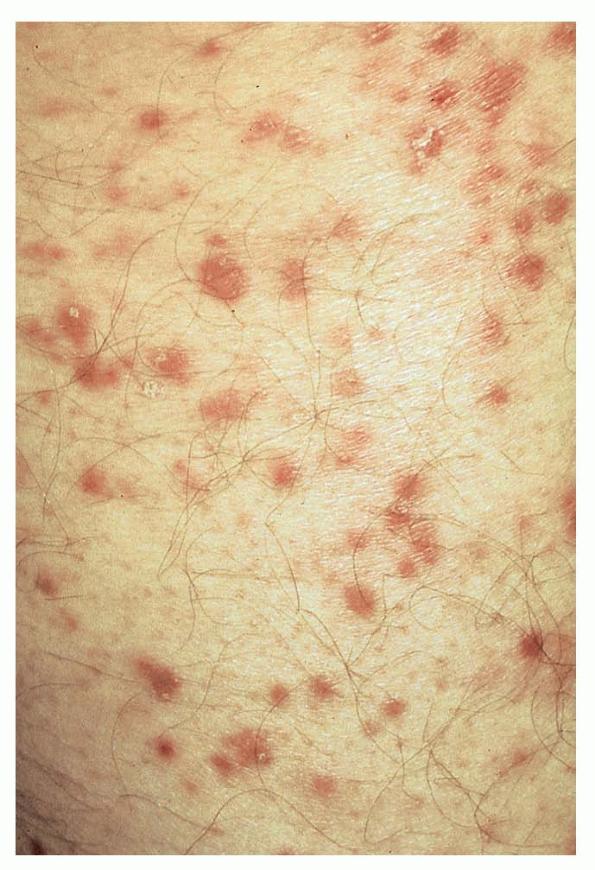
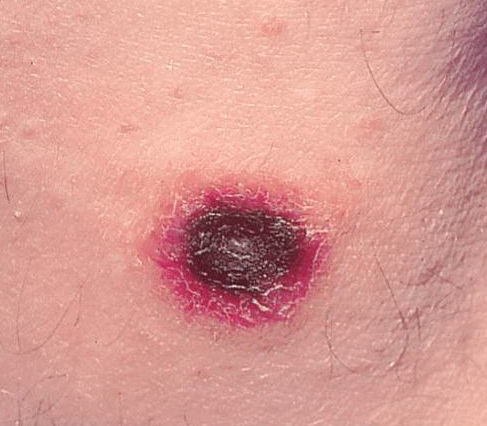
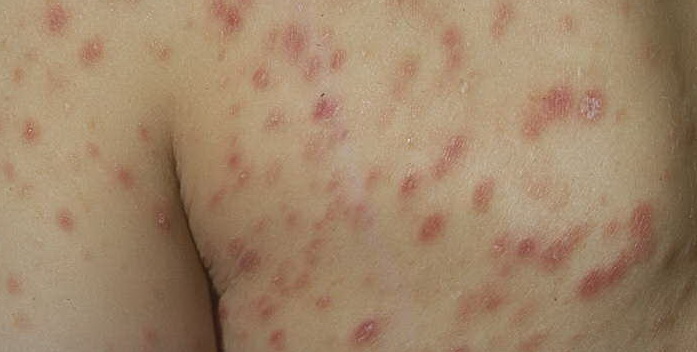
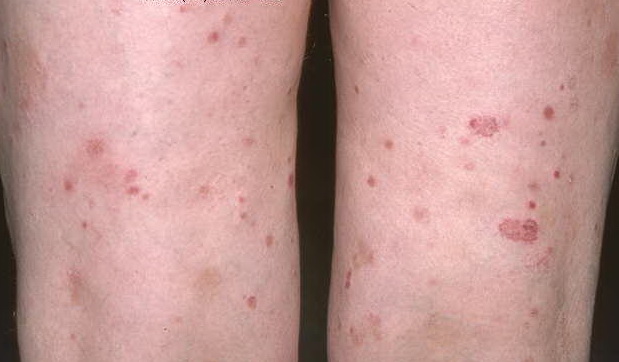
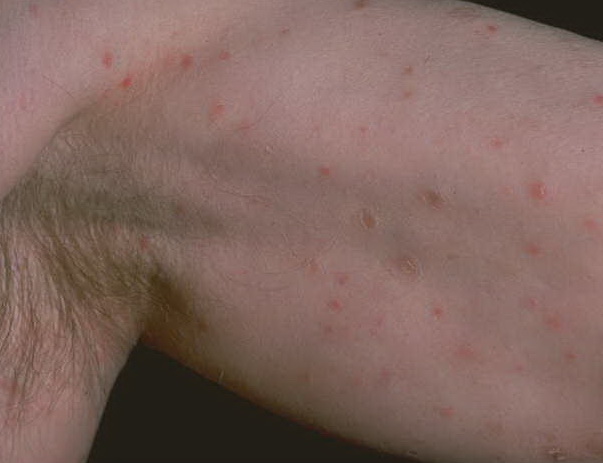
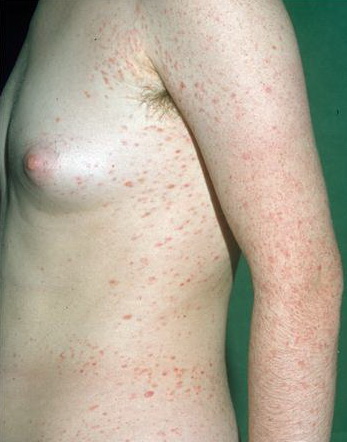
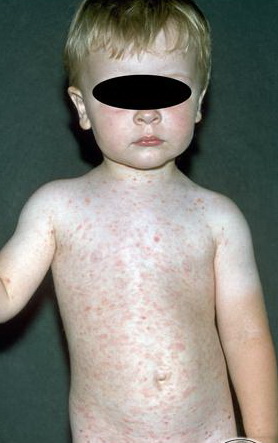
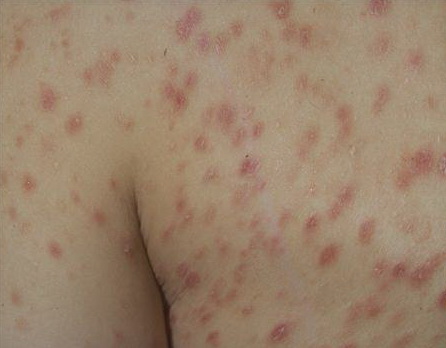
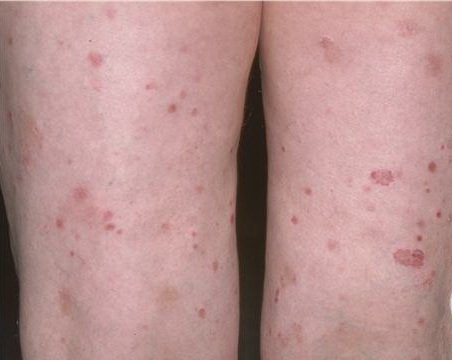
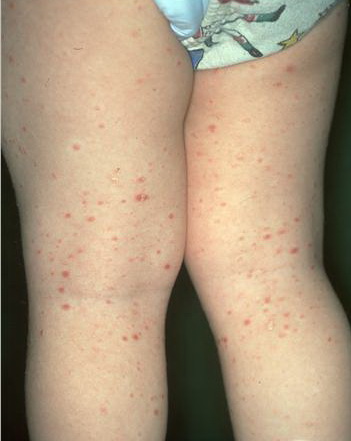
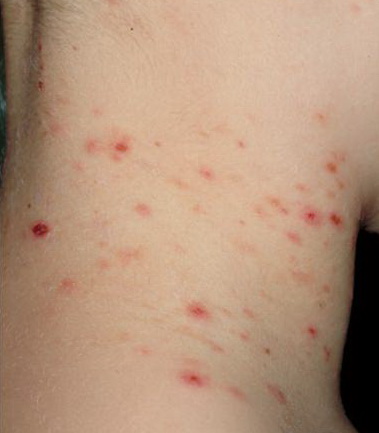
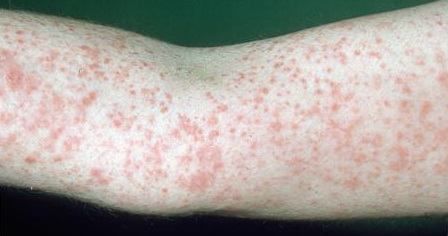
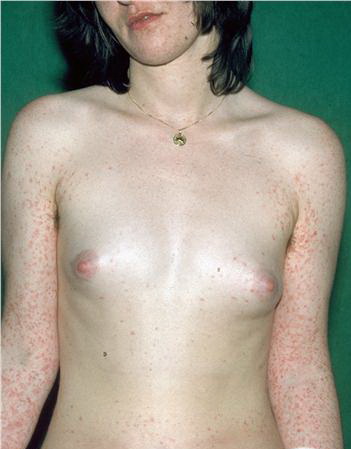
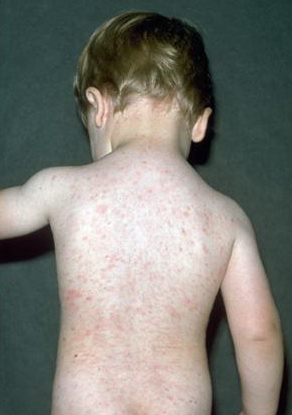
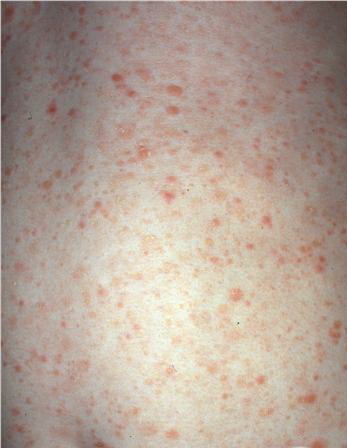
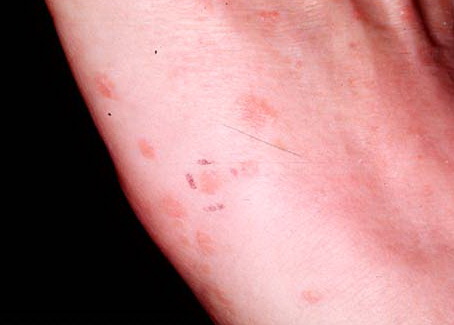
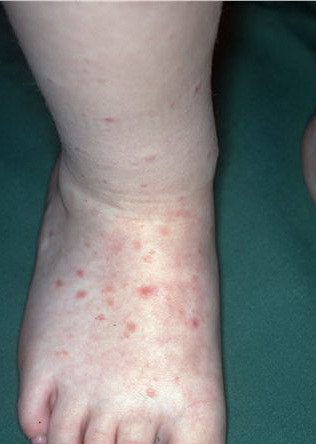
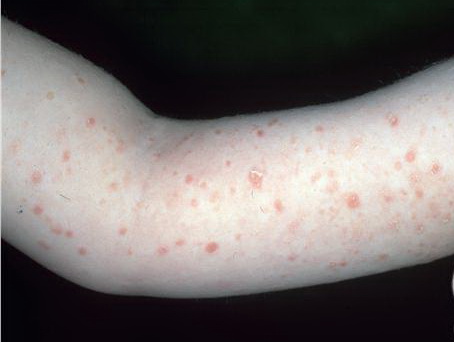
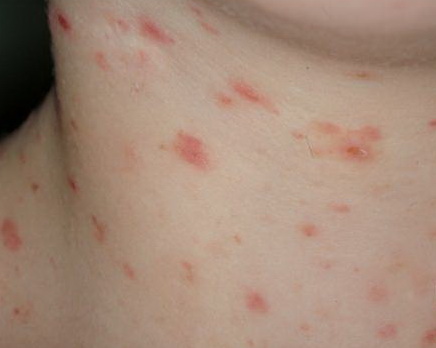
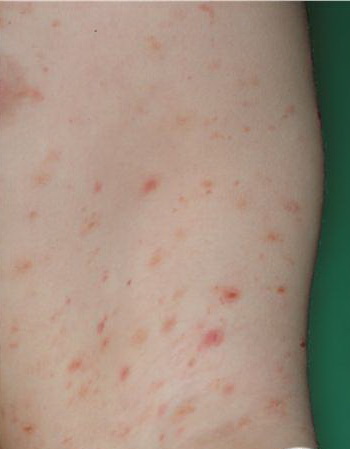
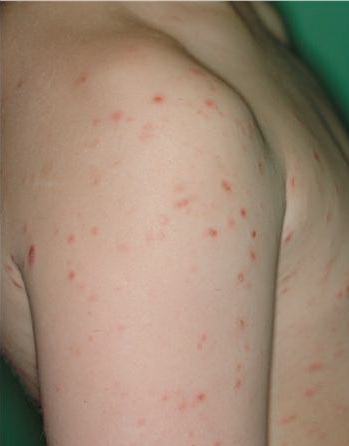
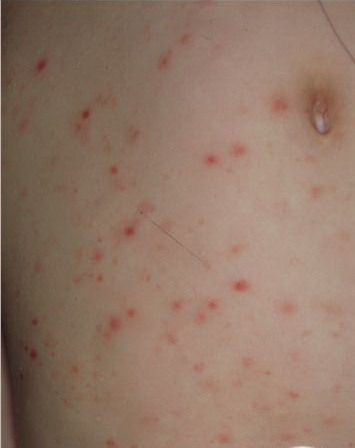
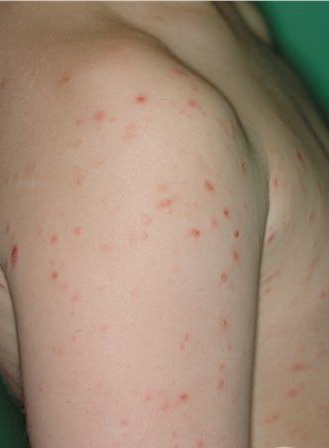
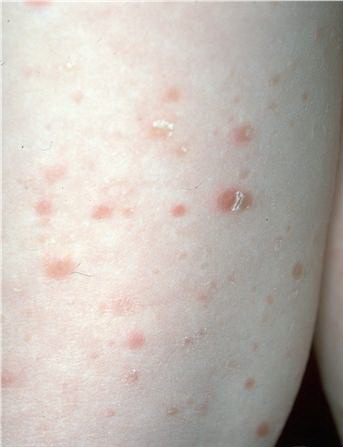
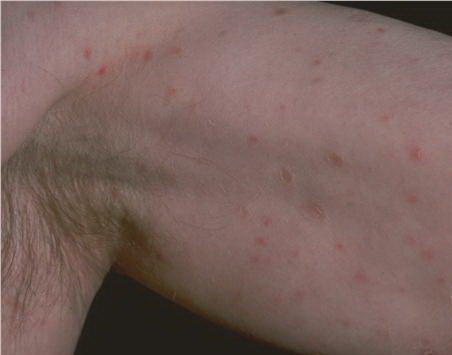
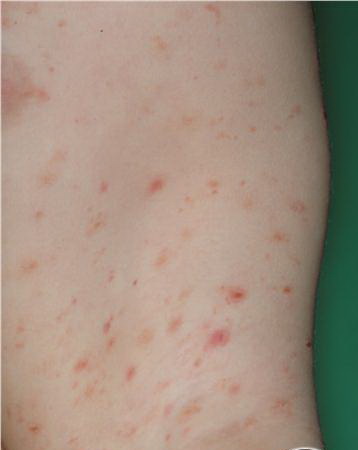
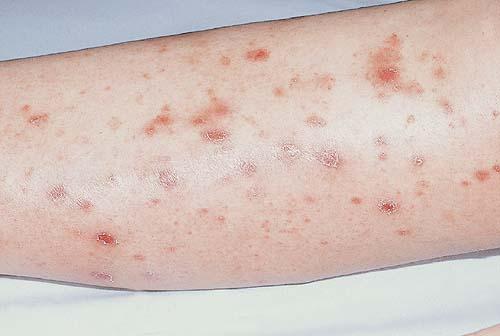
Lesions are often asymptomatic but can be pruritic or burning, especially in the more acute cases. PLC typically presents as recurrent crops of erythematous scaly papules that spontaneously regress over several weeks to months (Fig. 25-10). PLEVA manifests as recurrent crops of erythematous papules that develop crusts, vesicles, pustules, or erosions before spontaneously regressing within a matter of weeks . The more severe ulcerative variant is known as pityriasis lichenoides with ulceronecrosis and hyperthermia (PLUH) or febrile ulceronecrotic Mucha-Habermann disease. It presents as purpuric papulonodules with central ulcers up to a few centimeters in diameter . Some have proposed that this severe variant is actually an overt T-cell lymphoma.75 Pityriasis lichenoides lesions tend to concentrate on the trunk and proximal extremities,
but any region of the skin and even mucous membranes can be involved. Rare regional or segmental lesion distributions have been described. Although there are usually numerous co-existent lesions, occasionally only a small number of lesions will be present at any one time. All forms of pityriasis lichenoides can result in post-inflammatory hypopigmentation or hyperpigmentation. Chronic lesions can resolve with post-inflammatory hypopigmentation, sometimes presenting as idiopathic guttate hypomelanosis. Chronic lesions rarely lead to scars. In contrast, acute lesions result in deeper dermal injury and consequently often resolve leaving varioliform (smallpox-like) scars. The presence of lesions in various stages of evolution imparts a polymorphous appearance that is characteristic of pityriasis lichenoides.
Laboratory Tests
Miscellaneous non-specific abnormalities in blood test results occur but are of little practical value. Leukocytosis and a decreased CD4/CD8 ratio can occur.
HISTOPATHOLOGY
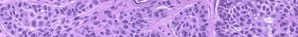
As with the morphology of the clinical lesions, pityriasis lichenoides can exhibit a range of histopathologic features encompassing acute, chronic, and intermediate lesional variants . All cases of pityriasis lichenoides contain an interface dermatitis that is denser and more wedge shaped in the acute lesions. The infiltrate is composed mainly of lymphocytes with a variable admixture of neutrophils and histiocytes. There is exocytosis, parakeratosis, and extravasation of erythrocytes. Epidermal damage ranges from intercellular and extracellular edema in less severe cases to extensive keratinocyte necrosis, vesicles, pustules, and ulcers. The acute variants can exhibit lymphocytic vasculitis with fibrinoid degeneration of blood vessel walls.
Occasional CD30+ lymphoid cells and occasional atypical lymphoid cells may be seen as a non-specific finding in many cutaneous lymphoid infiltrates. The presence of an appreciable numbers of these cells is not consistent with classic pityriasis lichenoides of any type and should raise concern for the lymphomatoid papulosis-CD30+ anaplastic large cell lymphoma disease spectrum.74 Other immunohistologic features and the clonality of pityriasis lichenoides are discussed in Etiology and Pathogenesis.
Differential Diagnosis
The differential diagnosis of pityriasis lichenoides includes many papular eruptions . Those that develop crusts, vesicles, pustules, or ulcers are grouped with PLEVA, whereas those that form predominantly scaly papules are grouped with PLC. Most of them can be excluded based on history and typical clinicopathologic features. A few, such as secondary syphilis and viral-associated lesions, also can be excluded based on serologic tests. Among the most challenging diseases to distinguish from pityriasis lichenoides are lymphomatoid papulosis and macular or papular variants of MF. As detailed earlier, the presence of large atypical lymphoid cells (often CD30+) differentiates lymphomatoid papulosis from pityriasis lichenoides.Macular or papular variants of MF are rare. They exhibit classic features of MF, including small atypical epidermotropic lymphoid cells with convoluted nuclei and a band-like superficial dermal lymphoid infiltrate.
Complications
Secondary infection is the most common complication of pityriasis lichenoides. PLEVA may be associated with low-grade fever, malaise, headache, and arthralgia. Patients with PLUH can develop high fever, malaise, myalgia, arthralgia, and gastrointestinal and central nervous system symptoms. Occasionally, debilitated patients may die. PLC has been associated uncommonly with LPP in children.79 Despite
their sometimes dominant T-cell clonal nature, PLC and PLEVA are considered clinically benign disorders without significant linkage to lymphomas or other malignancies.
Box 25-4 Differential Diagnosis of Pityriasis Lichenoides et Varioliformis Acuta (PLEVA) and Pityriasis Lichenoides Chronica (PLC)Most Likely
- o Arthropod bites, stings, infestations
- o Leukocytoclastic vasculitis
- o
- o Pityriasis rosea
- o Drug eruption
- o Guttate psoriasis
Consider
- o Folliculitis
- o Rickettsiosis
- o Erythema multiforme
- o Dermatitis herpetiformis
- o Spongiotic dermatitis, papular variant
- o Small-plaque parapsoriasis
- o Lichen planus
- o Gianotti-Crosti syndrome
Always Rule Out
- o Lymphomatoid papulosis
- o Secondary syphilis
- o Lymphomatoid papulosis
- o Mycosis fungoides (papular variant)
- o Secondary syphilis
Prognosis and Clinical Course
Pityriasis lichenoides has a variable clinical course characterized by recurrent crops of lesions that spontaneously resolve. The disorder may resolve spontaneously within a few months or, less commonly, persist for years. PLEVA usually has a shorter duration than PLC. Although the conclusion was not confirmed by subsequent investigation, one report suggested that the duration of pityriasis lichenoides in children correlated better with its clinical distribution than with the relative abundance of acute and chronic lesions, which often co-existed.59 From longest to shortest duration, the distribution of lesions ranged from peripheral (distal extremities) to central (trunk) to diffuse.
Box 25-5 Treatment of Pityriasis Lichenoides et Varioliformis Acuta and Pityriasis Lichenoides Chronica
FIRST LINE
- · Topical corticosteroids
- · Antibiotics (erythromycin 500 mg PO 2-4× daily80; tetracycline 500 mg PO 2-4× daily,81 minocycline 100 mg PO twice daily)
- · Phototherapy (sunbathing, ultraviolet B,82 ultraviolet A + ultraviolet B,83 narrowband ultraviolet B84)
SECOND LINE
- · Topical tacrolimus85,86
- · Prednisone (60/40/20 mg PO taper, 5 days each)87
- · Methotrexate (10-25 mg PO weekly)88,89
- · Phototherapy (ultraviolet AI, psoralen plus ultraviolet A)
- ·
- ·
Treatment
The mainstay of traditional therapy has been a combination of topical corticosteroids and phototherapy . Systemic antibiotics in the tetracycline and erythromycin families are used primarily for their anti-inflammatory rather than antibiotic effects. Cases with minimal disease activity may not require any treatment. The more acute the clinical course and the more severe the individual lesions, the more systemic therapy is indicated. Methotrexate is often effective in relatively low doses. Calcineurin inhibitors and retinoids may also be beneficial. Severe cases of PLEVA and PLUH often require systemic corticosteroids or similar drugs to gain control of systemic symptoms. Topical and systemic antibiotics may be needed to treat secondary infections complicating ulcerated skin lesions. These agents are often selected initially to cover Gram-positive pathogens, but subsequent use should be guided by culture results.
Prevention
There are no known preventive measures.