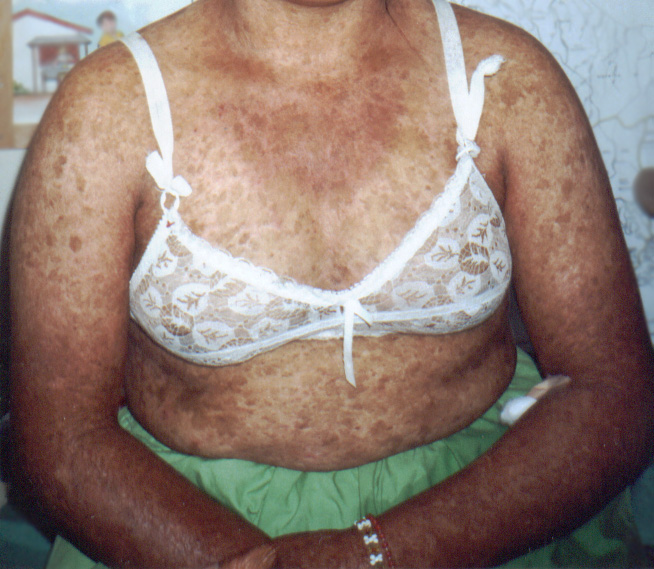
| Kindler syndrome =congenital acral skin blistering, photosensitivity, progressive poikiloderma, and diffuse cutaneous atrophy |
|
|
Kindler Syndrome
First described in 1954 by Theresa Kindler, Kindler syndrome is a rare autosomal recessive genodermatosis characterized by congenital acral skin blistering, photosensitivity, progressive poikiloderma, and diffuse cutaneous atrophy. The syndrome is a combination of features of inherited blistering skin disorders (eg, dystrophic epidermolysis bullosa) and congenital poikilodermas (eg, Rothmund-Thompson syndrome). Kindler syndrome is identified as entry 173650 in the Online Mendelian Inheritance of Man database. In 2003, Siegel et al mapped the disease locus to band 20p12.3 by using linkage and homozygosity analysis in an isolated cohort of patients with Kindler syndrome. Loss-of-function mutations were identified in the candidate gene FLJ20116, which was renamed KIND1. This gene encodes a 677–amino acid protein, kindlin-1, which is thought to play a regulatory role in inhibiting oversecretion of basement membrane components by basal keratinocytes at the dermoepidermal junction.2,3
Kindlin-1 is a human homolog of the Caenorhabditis elegans protein UNC-112, a membrane-associated structural/signaling protein that had been implicated in linking the actin cytoskeleton to the extracellular matrix (ECM). Kindler syndrome is the first genodermatosis caused by a defect in actin-ECM linkage rather than keratin-ECM linkage, underlying the pathology of other inherited skin fragility disorders such as epidermolysis bullosa. Since the first description in 1954 by Theresa Kindler, more than 100 cases of Kindler syndrome have been reported worldwide. A cluster of 26 patients with the syndrome has been identified within a tribe in the Bocas del Toro province on the northwestern Caribbean coast of Morbidity and mortality are mostly related to secondary infections arising from cutaneous bullae and to cosmetic disfigurement. Persons of any race can be affected. No sex predilection has been documented. Patients usually present with the initial skin manifestations during the first year of life.
Kindler syndrome has been shown to result from mutations in the KIND1 gene on band 20p12.3 (see Pathophysiology). An autosomal recessive pattern of transmission has been reported, but sporadic cases are common, with many originating in consanguineous families. Variable expressivity within families has also been documented. Mutations in the gene encoding type VII collagen (COLA7A1) have been excluded, distinguishing Kindler syndrome from dystrophic epidermolysis bullosa Treatment Treatment is mainly symptomatic and preventative in nature. Patients should be advised to avoid trauma, which helps prevent blister formation. Sun avoidance and photoprotection may prevent or slow the progression of poikiloderma. Good wound care includes the use of topical and systemic antibiotics for infected bullous lesions, which may reduce morbidity. Telangiectasias can be treated with pulsed-dye laser therapy. Surgical correction of urethral, anal, or esophageal stenosis may be needed. Consultations
The role of pharmacotherapy is supportive in nature. Emollients and good blister care reduce morbidity and help prevent complications.
|