Albinism -and Other
Genetic Disorders of
Pigmentation
Epidemiology of Albinism
Oculocutaneous albinism (OCA) is the most common inherited disorder of generalized hypopigmentation, with an estimated frequency of 1 in 20,000 in most populations. Four different types of OCA have been described. OCA types 1 and 2 (OCA1, OCA2) are most frequent and account for approximately 40 percent and 50 percent, respectively, of OCA cases worldwide. OCA2 occurs in approximately 1 in 30,000 to 1 in 36,000 Caucasians and 1 in 10,000 to 1 in 17,000 blacks in the United States,1-3 but is reported at higher frequencies ranging from 1 in 1400 to 1 in 7000 in sub-Saharan Africa.4,5 OCA3 and OCA4 are far less frequent, although the rufous OCA phenotype, described later, associated with OCA3 in southern African blacks has been reported at an incidence of approximately 1 in 8500.
Hermansky-Pudlak syndrome (HPS) is rare except in the Caribbean island of Puerto Rico, particularly in the northwestern region where the majority of patients are found, and has an incidence there of 1 in 1800.7 Chédiak-Higashi syndrome (CHS) is also quite rare.
Epidemiology of Congenital Disorders of Pigmentation
Waardenburg syndrome (WS) is probably less frequent than OCA. The highest reported incidence is 1 in 20,000 in Kenya, and most estimates of its incidence in the Netherlands, where it was originally reported, are in the range of 1 in 40,000. Incidences of WS with deafness are lower, ranging between 1 in 50,000 and 1 in 212,000. WS has been described occurring at varying frequencies in the congenitally deaf, ranging from 0.9 percent to 2.8 percent in some studies to 2 percent to 5 percent in others. The incidence of piebaldism
is estimated to be lower than 1 in 20,000.
ALBINISM AND CONGENITAL
DISORDERS OF
PIGMENTATION AT A GLANCE
- World-wide occurrence.
- Typically inherited with marked differences in penetrance. Albinism is usually a recessive trait, and congenital disorders of pigmentation usually show a dominant pattern of inheritance.
- Clinical features of albinism include lightly pigmented or non-pigmented skin and silvery white or lightened hair color.
- Clinical features of congenital disorders of pigmentation include patches of white hair (poliosis), variations in iris color, and depigmented patches of white skin.
- Ocular nystagmus and reduced visual acuity distinguish albinism from congenital disorders of pigmentation.
- In albinism melanocytes have reduced or absent DOPA positivity. In certain congenital disorders of pigmentation, affected areas of skin can lack melanocytes completely.
▪
ETIOLOGY, PATHOGENESIS, AND
CLINICAL FEATURES
Awareness of the biologic basis of the distinction between congenital disorders of pigmentation, which are disorders of melanocyte development, and the varieties of albinism, which are disorders of melanocyte differentiation, can be important for fully understanding their clinical manifestations. Albinism results from the dysfunction of a normal complement of pigment cells, which results in complete or partial loss of cutaneous pigmentation. The forms of albinism, including the sub-types of OCA as well as albinism syndromes with systemic manifestations, result either from enzymatic defects in the biosynthesis of melanin, from melanosomal defects that interfere with melanin formation, or from problems in the intracellular transport and localization of proteins essential for melanin biosynthesis . Congenital disorders of pigmentation usually result from mutations in genes critical for melanocyte development during embryogenesis. These disorders can also be associated with other systemic problems because of the requirement for these gene products in the development of cell types other than melanocytes. More accurate descriptors of these disorders could be congenital (or genetic) disorders of melanocyte differentiation and congenital (or genetic) disorders of melanocyte development, to reflect the fact that both categories of conditions, albinism and developmental pigmentary syndromes, are generally inherited but result from different mechanisms of disease .
Etiology, Pathogenesis, and Clinical Features of Albinism
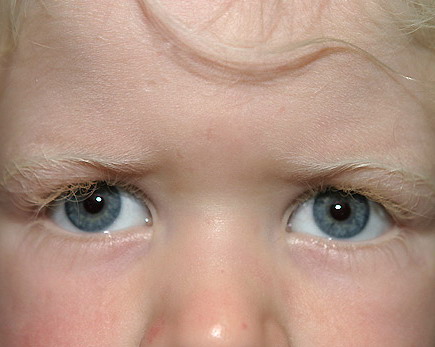
Although the pigmentary abnormalities associated with various types of albinism can vary widely, common to all types of albinism is reduced visual acuity and ocular nystagmus, a result of misrouting of the optic nerve at the optic chiasm and foveal hypoplasia. This has been described not only in humans10 but also in other albino mammals.11 Experiments using the tyrosinase promoter to express both tyrosinase and tyrosine hydroxylase in albino transgenic mice suggest that the tyrosine hydroxylase activity of tyrosinase is particularly important for ensuring the proper routing of retinal projections at the optic chiasm during development.12,13 The ocular manifestations of albinism can vary greatly, ranging from severe (legal blindness) to nearly undetectable. They also include a reduction in iris pigment, a reduction in retinal pigment, and alternating strabismus.
OCULOCUTANEOUS
ALBINISM TYPE 1
OCA type 1 [OCA1; Online Mendelian Inheritance in Man (OMIM) #203100] is produced by loss of function of the melanocytic enzyme tyrosinase resulting from mutations of the TYR gene. Null mutations are associated with a total loss of function and no pigment formation (OCA1A), whereas “leaky” mutations result in an enzyme that retains some function and is associated with some pigment formation (OCA1B).
Analyses of DNA from individuals with OCA1A have shown a large number of different mutations in the TYR gene. These mutations include missense, nonsense, frameshift, splice site, and deletion mutations. Most individuals with OCA1 are compound heterozygotes with different mutant maternal and paternal alleles.6 Missense mutations in the TYR gene are distributed among distinct regions of the coding sequence, which suggests that the encoded protein has multiple functional domains. Two of the clusters are in the copper-binding regions, with a third near the amino terminus of the mature protein, in the extra-melanosomal domain of tyrosine shown to require phosphorylation for enzyme activation.17,18 Clustering of mutations in discrete regions of the coding sequence is consistent with these regions' being important either for the melanogenic activity of tyrosinase or for functions related to its maturation or processing.6,18 Missense mutations at the signal peptide cleavage site19,20 implicate this cleavage event as an important step in the development of tyrosinase activity. Frameshift mutations near the C-terminus of the coding region21,22 indicate that the cytoplasmic domain of tyrosinase is also important for full activity, possibly because of the presence of protein kinase C-β-dependent phosphorylation sites that have been identified at the extreme C-terminus of the protein.
All nonsense and frameshift mutations are associated with a complete loss of tyrosinase activity, presumably because of the subsequent production of a truncated protein. With missense mutations the picture is more complicated. A set of missense mutations that were associated with the accumulation of pigment with age in either OCA1B or temperature-sensitive OCA (OCA1TS) patients23 were shown to have residual enzymatic activity. Hence, it is likely that a subset of TYR missense mutations are responsible for the OCA1B and OCA1TS phenotypes because of the reduced, rather than absent, tyrosinase activity in their melanocytes.6 However, other missense mutations leading to the OCA1A and OCA1TS phenotypes result in defective intracellular processing of tyrosinase and retention of the mutant tyrosinase proteins in the endoplasmic reticulum, which suggests that some molecular variants of OCA1 represent an endoplasmic reticulum retention disease.24-26 Thus, the residual enzymatic activity of a missense tyrosinase mutant cannot fully predict its phenotype, because other, presumably
conformational, determinants of nascent mutant proteins may lead to their retention in the endoplasmic reticulum, block transport to the melanosome, and cause a more severe pigmentary phenotype.
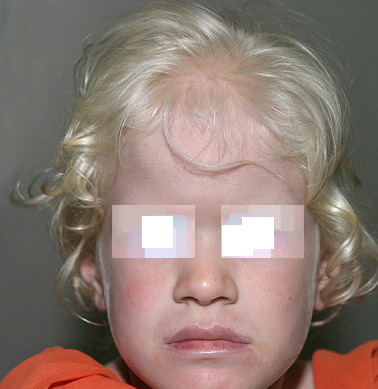
In OCA1A, or the classic tyrosinasenegative OCA, there is a complete inability to synthesize melanin in skin, hair, and eyes, which results in the characteristic “albino” phenotype. Affected individuals are born with white hair and skin, and there are no changes as they mature. The phenotype is the same in all ethnic groups and at all ages. The hair may develop a slight yellow tint due to denaturing of the hair protein related to sun exposure and/or shampoo use. The irides are translucent, appear pink early in life, and often turn a gray-blue color with time. No pigmented lesions develop in the skin, although amelanotic nevi can be present. The architecture of skin and hair bulb melanocytes is normal. The melanosomes show a normal melanosomal membrane, and normal internal matrix formation is observed in stage 1 and 2 melanosomes.
The phenotype of OCA1B can range from minimal hair pigment to near-normal skin and hair pigment. Most individuals with OCA1B have very little or no pigment at birth and develop varying amounts of melanin in the hair and skin in the first or second decade of life . In some cases the melanin develops within the first year. The hair color changes to light yellow, light blond, or golden blond first, as a result of residual pheomelanin synthesis, and eventually can turn dark blond or brown in adolescents and adults. The irides can develop light tan or brown pigment, sometimes limited to the inner third of the iris, and iris pigment can be present on globe transillumination. However, some degree of iris translucency, as demonstrated by slit-lamp examination, is usually present. Many individuals with OCA1B will tan with sun exposure, although it is more common to burn without tanning. Pigmented lesions (nevi, freckles, lentigines) develop in the skin of individuals who have developed pigmented hair and skin. In some patients, the moderate amount of residual tyrosinase activity can lead to near-normal cutaneous pigmentation, so that the clinician may overlook subtle cutaneous pigmentary abnormalities and render instead the mistaken diagnosis of ocular albinism.
One variation of OCA1B is the temperature-sensitive phenotype. In this variation, scalp and axillary hair remain white or slightly yellow, but arm and leg hair pigments. The skin remains white and does not tan. The retention of melanin
synthesis in the cooler areas of the body, such as the arms and legs, but not the warmer areas, such as the trunk and the scalp, is associated with a temperature-sensitive mutation in tyrosinase, which loses activity above 35°C (95°F).23 Similar tyrosinase mutations have been described in the Himalayan mouse27 and in the Siamese (pointed) cat.
OCULOCUTANEOUS
ALBINISM TYPE 2
Mutations of the P gene, which maps to chromosome arm 15q, are responsible for OCA2 (OMIM #203200).29 OCA2 occurs worldwide, although somewhat more frequently in the African, African American, and certain Native American populations. Historically, affected individuals have benefitted from limiting their sun exposure, especially in desert and equatorial climates. Interesting anthropologic studies have described how various societies have differed in their treatment of members with OCA2. From the standpoint of melanin synthesis, the defect in OCA2 appears to involve primarily a reduction in eumelanin synthesis, with less effect on pheomelanin synthesis. The predicted structure of the P gene, a melanosomal protein, contains 12 transmembrane domains.31,32 As expected, a number of mutations of the human P gene are associated with human OCA2.
In sub-Saharan Africa, a single 2.7-kb deletion allele accounts for 60 percent to 90 percent of mutant P alleles and is associated with a common haplotype, which suggests a common founder.5,34-36 It has been estimated that this single mutation is associated with 25 percent to 50 percent of all mutant P alleles in African Americans.5,34-40 However, other diverse mutant alleles have been described in this population and in Africans. The Brandywine, Maryland, isolate is an inbred American population, originally located in a rural area east of Washington, D.C., that has been studied extensively for its prevalence of albinism, dentinogenesis imperfecta, and osteogenesis imperfecta, with mixed Caucasian, African, and possibly Native American ancestry.41 In this isolate, 1 in 85 individuals has OCA241,42 and is homozygous for the 2.7-kb deletion allele of the P gene. Thus it is likely that this 2.7-kb deletion allele accounts for the distinct OCA2 phenotype in Africans and African Americans.
OCA2 also has been reported at relatively high frequencies ranging from 1 in 28 to 1 in 6500 in specific Native American groups, including those populations in the southwest United States (Hopi population), southern Mexico, eastern Panama (Cuna population), and southwest Brazil.30 In the Navajo population, a homozygous 122.5-kb deletion has been described in members with OCA2. This mutation results in the loss of exons 10 to 20 of the P gene, corresponding to a region containing seven of the transmembrane domains, and appears to be specific for OCA2 within the Navajo population.44 Unlike the mutations in TYR, the missense mutations described to date in the P gene do not seem to cluster in any specific region.
Regarding P gene product function, it has been shown that melanosomes from p protein-deficient melanocytes have an abnormal pH. Melanosomes in cultured melanocytes derived from wild-type mice are typically acidic, whereas melanosomes from p protein-deficient mice are nonacidic.45 Hence, it is likely that the p protein regulates the acidic pH of melanosomes, perhaps by functioning as an anion co-transporter in conjunction with a proton pump on the melanosomal membrane. An alternate possibility is that the acidic conditions mediated by the p protein favor the normal biogenesis of melanosomes, including the correct targeting of other melanosomal proteins such as tyrosinase.
In African and African American individuals, there is a distinct OCA2 phenotype . Hair is yellow at birth and remains so throughout life, although the color may turn darker. Hair color can turn lighter in older individuals, and this probably represents normal graying with age. The skin is creamy white at birth and changes little with time. No generalized skin pigment is present, and no tan develops with sun exposure, but pigmented nevi, lentigines, and freckles often develop. The irides are blue-gray or light tan or brown. The development of lentigines or ephelides, well-demarcated pigmented patches usually on sun-exposed areas of the skin, may be evidence of a separate genetic susceptibility, because these lesions develop only in some OCA2 families and not in others. The presence of ephelides is associated with a lower risk of skin cancer in South African individuals.
The brown OCA phenotype is a distinct OCA2 phenotype that has been described in the African and African American populations. In this clinically less severe phenotype,48 the hair and skin are light brown and the irides are gray to tan at birth
. With time, skin color changes little, but the hair may turn darker, and the irides may accumulate more tan pigment. The skin generally does not burn but may darken with sun exposure. Affected individuals are recognized as having albinism rather than a variation in normal pigmentation because of the ocular changes present. The iris has punctate and radial translucency, and moderate retinal pigment is present. Visual acuity ranges from 20/60 to 20/150. In brown OCA, the amount of eumelanin in the skin and hair is reduced but not absent. Studies have now shown that brown OCA is associated with heterozygosity for P gene alleles, one of which is null and the other partially functional
In Caucasian individuals with OCA2, the amount of hair pigment present at birth or developing with time varies from minimal in northern Europeans (particularly Scandinavians) to moderate in southern European or Mediterranean individuals. The hair can be very lightly pigmented at birth, having a light yellow or blond color, or more pigmented with a definite blond, golden blond, or even red color. The normal delayed maturation of the pigment system and sparse hair early in life can make it difficult to recognize albinism early in normally pigmented northern European individuals. For all types of OCA in northern European families, the cutaneous hypopigmentation at birth or early in life is often similar to that of the parents and relatives, and concern is only raised when it appears that the child is not tracking well visually or has developed nystagmus.
The skin is creamy white and does not tan. The iris is blue-gray or lightly pigmented, and the amount of translucency
correlates with the development of iris pigment. With time, pigmented nevi and lentigines may develop, and pigmented freckles are seen in areas with repeated sun exposure. The hair in Caucasian individuals may slowly turn darker through the first two or more decades of life.
PRADER-WILLI AND
ANGELMAN
SYNDROMES
Prader-Willi and Angelman syndromes often are associated with hypopigmentation.50,51 The intragenic deletion encompassing one P allele in these patients52,53 suggests that the observed pigmentary phenotype is related to OCA2 and the P gene, even if the details of this association are not fully understood.
OCULOCUTANEOUS
ALBINISM TYPE 3
Four distinct sets of mutations in the TYRP1 gene resulting in OCA3 (OMIM #203290) have now been described. The first mutation was found in an African American newborn twin who was initially classified clinically as having brown OCA. Mutation analysis revealed a single-base deletion at codon 368 producing a frameshift and premature stop codon in exon 6 and a slightly truncated TYRP1 molecule.57 This mutation is shared by a substantial proportion of the rufous OCA population in southern Africa.58 Rufous OCA is a distinct OCA phenotype in which the skin color is a mahogany brown with a slight reddish hue, and the hair color varies from deep mahogany to sandy red.2,58 The second TYRP1 mutation, also identified in the rufous OCA population, is a single-base substitution at codon 166 resulting in the alteration of a serine to a premature stop codon in exon 3 and a truncated TYRP1 molecule.58 In a Pakistani kindred, individuals homozygous for another premature termination mutation have been described.59 A Caucasian male was compound heterozygous for a missense mutation, inherited from the patient's mother, in TYRP1 located in the second copper-binding domain and a stop codon, which apparently occurred spontaneously.
OCA3 has presented with both the brown OCA and the rufous OCA phenotypes in the African and African American populations. In the two examples of individuals not of African descent, the phenotype has been that of a tyrosinase-positive albinism, such as OCA1B or OCA2. As additional examples of OCA3 are characterized, a genotype-phenotype correlation may become clearer.
OCULOCUTANEOUS
ALBINISM TYPE 4
OCA4 (OMIM #606574) appears rarely worldwide but is more common in East Asian populations. A variety of mutations, including a splice acceptor site mutation and missense mutations, have been found in a gene called MATP (membrane-associated transporter protein), located on chromosome arm 5p. OCA4 can have a variable phenotype, ranging from absence of pigmentation to some pigmentation with brown irides. Improvement during the first decade of life has been reported.
The protein product of the MATP gene is predicted to be a membrane protein spanning the membrane 12 times and containing a conserved sucrose transporter signature sequence, which suggests an important functional role for this motif.64 In addition, melanosome anomalies have been observed in the medaka fish and mouse homologs of OCA4These data indicate that MATP/Matp plays a critical role in vertebrate pigmentation and suggest that MATP/Matp may be a component of the melanosomal membrane, presumably mediating the transport of a molecule required for melanogenesis or for another melanosome function