Actinic Prurigo
EPIDEMIOLOGY
Actinic prurigo (AP) appears to occur throughout much of the world. Native Americans at all latitudes are particularly affected but also, more rarely, inhabitants of the United Kingdom, the United States, Europe, Australia, and Japan.
PATHOGENESIS
AP appears to be UVR induced, in that it is more severe in spring and summer and reasonably often demonstrates abnormal skin phototest responses to the UVB or UVA wavelengths, or both. In addition, sunlight exposure and solar simulating irradiation may sometimes induce a rash resembling PMLE, many patients have close relatives with PMLE,18 and a dermal, perivascular mononuclear cell infiltrate similar to that of PMLE may be seen in early lesions.
ACTINIC PRURIGO AT A GLANCE
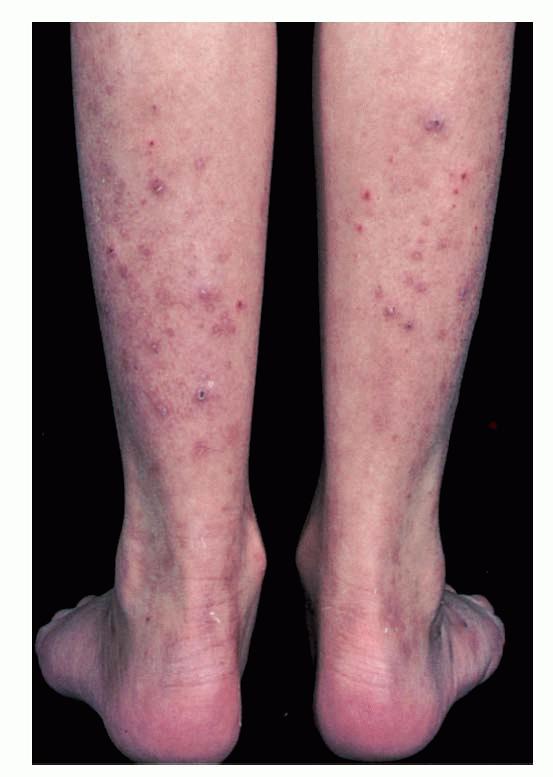
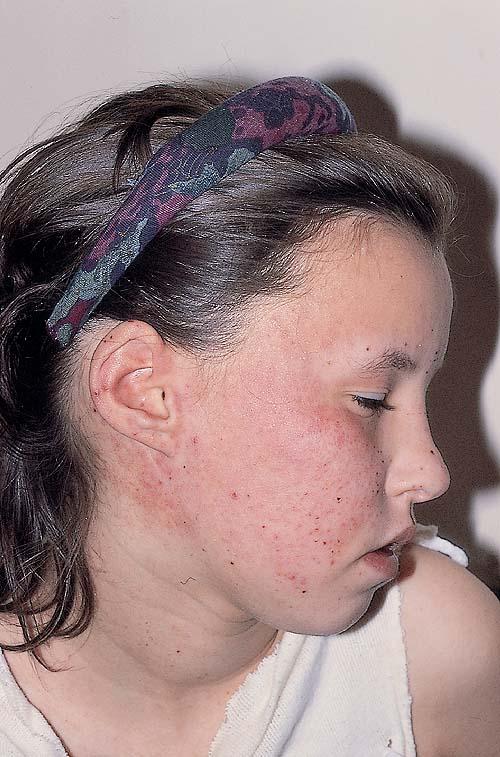
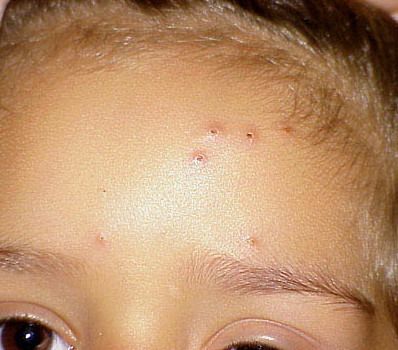
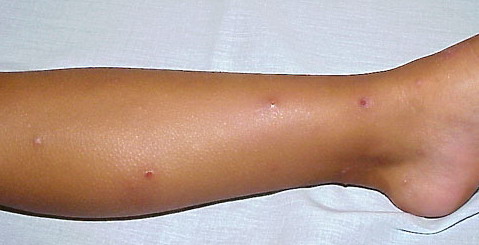
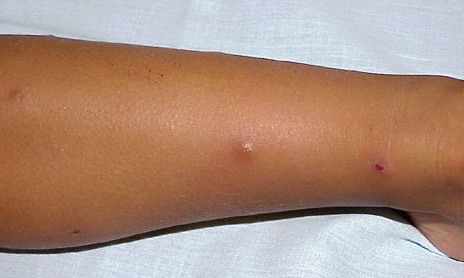
- A rare, persistent, pruritic, excoriated, papular, or nodular eruption of sun-exposed and, to a lesser extent, nonexposed skin.
- Usually begins in childhood, may remit at puberty, is generally worse in summer, and normally fades in winter.
- A persistent variant of the sometimes co-existent polymorphic light eruption (PMLE).
- Similar to hereditary or familial PMLE that predominantly affects native North, Central, or South Americans but that is usually more severe and generally persists into adulthood.
- In addition to avoidance of ultraviolet irradiation, often oral thalidomide or other immunosuppressive agent is required.
- Prophylactic immunosuppressive phototherapy and topical application of calcineurin inhibitors can perhaps reduce recurrences if the eruption is cleared first.
AP may therefore be a slowly evolving, excoriated form of PMLE, and thus also a DTH reaction. In addition, human leukocyte antigen (HLA) DRB1*0401 (DR4), which is present in approximately 30 percent of normal individuals, occurs in around 80 to 90 percent of those with AP, whereas HLA DRB1*0407, found in approximately 6 percent of normal individuals and not infrequently in Native Americans, occurs in around 60 percent, so this inherited feature may well be responsible for converting PMLE into AP. In addition, some patients with the AP tissue type demonstrate clinical PMLE but also have persistent lesions, whereas in some patients clinical AP converts to clinical PMLE and in others clinical PMLE changes to clinical AP, all of which further suggests a relationship between the
two disorders. The cutaneous molecular UVR absorbers responsible for initiating the eruption are not known but may well be diverse, as suggested for PMLE.
CLINICAL FEATURES
History.
AP is more common in females and usually begins by age 10 years. A positive family history of either AP or PMLE is present in about a fifth of patients, but the incidence of atopy, at around 10 percent, is not increased. The eruption is often present all year round, but is generally worse in summer, although very rarely it is worse in winter or both spring and fall, with immunologic tolerance presumably developing during the summer in these instances. Exacerbations tend to begin gradually during sunny weather rather than after specific sun exposure, although PMLE-like outbreaks are also possible.
Cutaneous Lesions.
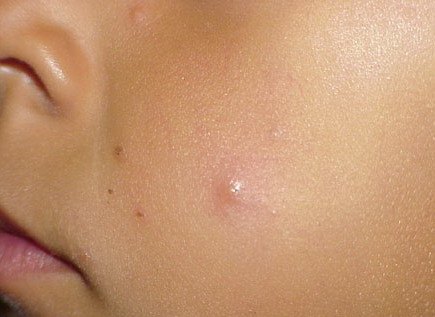
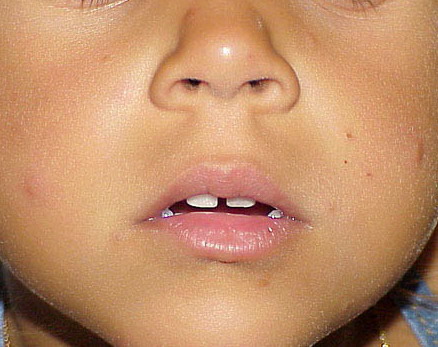
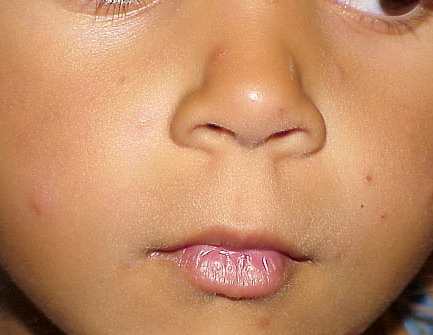
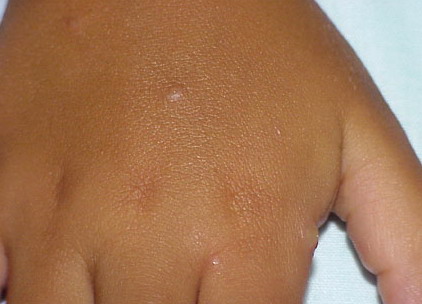
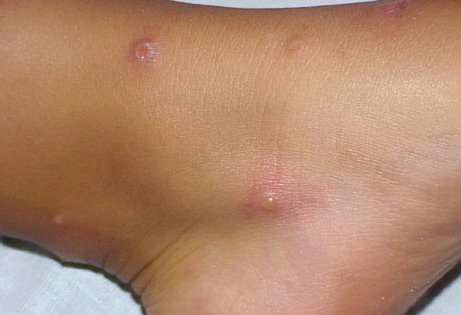
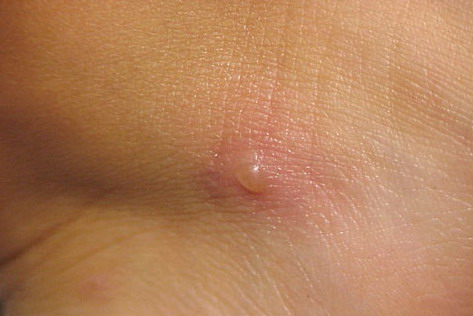
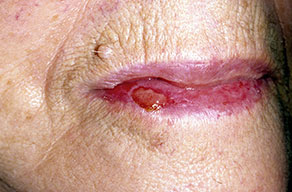
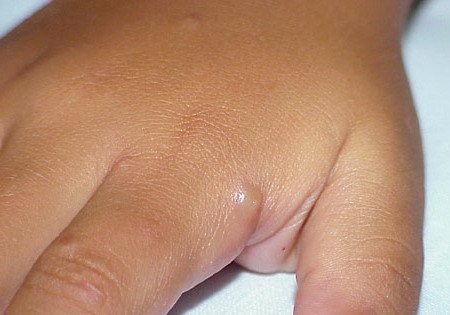
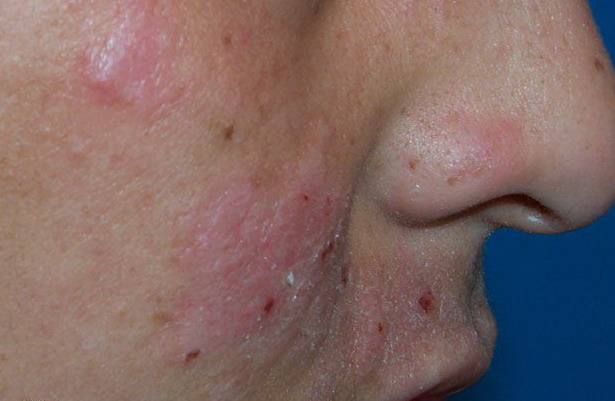
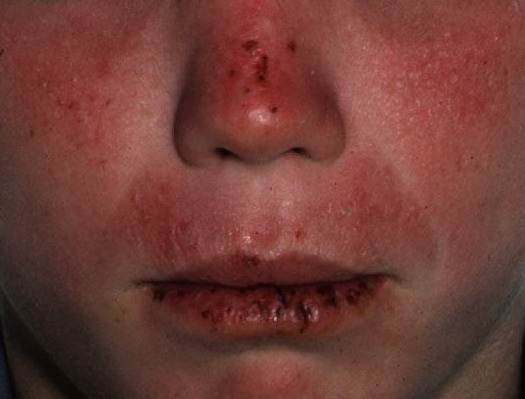
Lesions are typically very pruritic, usually excoriated papules or nodules , sometimes associated with variable eczematization, lichenification, or crusting. All exposed areas are usually affected, particularly consistently uncovered sites, with a gradual fading toward habitually covered skin. The latter areas are also often mildly affected, particularly over the sacral area and buttocks. Cheilitis, particularly of the lower lip, and conjunctivitis are also possible, particularly in Native Americans.Healed facial lesions may leave small pitted or linear shallow scars.
LABORATORY TESTS
Histology.
Early papular lesions show changes similar to those of PMLE, namely, mild acanthosis, exocytosis, and spongiosis in the epidermis and moderate lymphohistiocytic, dermal perivascular infiltration,23 even very rarely suggesting lymphoma. In persistent lesions, however, excoriations, increasing acanthosis, variable lichenification, and dense mononuclear cell infiltration lead to a non-specific appearance.
Box 90-2 Differential Diagnosis of Actinic Prurigo
Most Likely
- Polymorphic light eruption
- Atopic eczema
- Photo-exacerbated atopic or seborrheic eczema
- Insect bites
- Prurigo nodularis
Always Rule Out
Blood Tests.
Assessment of the circulating anti-nuclear and extractable nuclear antibody titers should normally be undertaken to exclude subacute cutaneous or other form of lupus. The finding of HLA type DRB1*0401 (DR4) or DRB1*0407, particularly the latter, supports the diagnosis of AP.
Phototesting.
Cutaneous phototesting with a monochromator confirms light sensitivity in up to half of cases36 but as in PMLE does not discriminate from other photodermatoses. Provocation testing with a solar simulator or other broadband sources may occasionally induce a PMLE-like eruption
DIFFERENTIAL DIAGNOSIS
.
COMPLICATIONS
Permanent mild scarring, particularly on the face, and small, circular, atrophic, usually hypopigmented lesions elsewhere may arise from the chronic excoriations usual in the disease.
PROGNOSIS AND CLINICAL COURSE
AP generally arises in childhood and often improves or resolves in adolescence, although persistence into adult life, sometimes in the form of PMLE, is possible. More rarely, the disorder arises in adulthood and persists indefinitely. A closely related condition, which may affect North, Central, or South American patients of Native American or mixed race, appears indistinguishable from AP
.
PREVENTION
Annual prophylactic phototherapy in spring or the use of the topical calcineurin inhibitors tacrolimus and pimecrolimus sometimes prevents expression of the established disease. There is no known way to prevent its initial onset.
TREATMENT
The restriction of sun exposure and use of broad-spectrum, high-protection-factor sunscreens may help in milder cases, assisted by intermittent use of topical or perhaps rarely oral steroids. Oral thalidomide, generally in low doses (50 to 100 mg at night) and preferably given intermittently, is almost always effective within weeks for more resistant disease in all age groups.42 Adverse effects are generally mild and may include drowsiness, headache, constipation, and weight gain, whereas careful studies of nerve conduction performed every few months are important to minimize the risk of slowly progressive, probably dose-related peripheral neuropathy. Pregnancy must also be rigorously avoided because of the high risk of teratogenic effects. If thalidomide is unavailable or unsuitable, phototherapy with narrowband UVB or PUVA may occasionally help, perhaps more reliably if the skin has been cleared first with oral steroids. Topical tacrolimus or pimecrolimus may also apparently help on occasion, again if the skin is cleared first, and oral immunosuppressive therapy with azathioprine or cyclosporine may also be useful if the other therapies are ineffective, unsuitable, or not tolerated. Treatment for hereditary AP is the same as for sporadic AP.