| Ataxia-telangiectasia= الهزع المخيخي مع توسع الشعريات |
|
|
Ataxia Telangiectasia
EPIDEMIOLOGY
Ataxia-telangiectasia , also called Louis-Bar syndrome, is an autosomal recessive disorder with an incidence of approximately 1:40,000 and a carrier rate of up to 1 percent. Carriers have an increased risk of breast cancer, hematologic malignancies, and ischemic heart disease, with a reduced life expectancy of approximately 8 years.111 These heterozygotes show increased risk of chromosomal breaks after exposure to irradiation in vitro, suggesting that mammograms in known carriers of AT are contraindicated.
ETIOLOGY AND PATHOGENESIS
AT results from mutations in ataxia-telangiectasia mutated (ATM), which encodes a phosphatidylinositol 3-kinase-like serine/threonine protein kinase that plays a central role in activating apoptotic and cell cycle responses to DNA damage.112 More than 400 different ATM mutations have been identified in patients with AT. The MRE11-RAD50-NBS1 (MRN) complex senses DNA breaks and recruits and activates ATM. The autophosphorylated ATM monomers then phosphorylate and thus activate several targets, among them p53, BRCA1, and NBS1 and MRE11 themselves, leading to cell cycle arrest and facilitated DNA repair. The activation of ATM occurs in response to external damage, such as from ionizing radiation and radiomimetic agents (bleomycin), and in physiologic DNA breaks, such as during meiosis, telomere maintenance, and V(D)J recombination in lymphocytes. These important roles of ATM explain the immunodeficiency, premature aging, progressive neurologic deterioration, and sensitivity to ionizing radiation. Oxidative stress related to ATM dysfunction has also been implicated.
ATAXIA-TELANGIECTASIA AT AGLANCE
Mutations in the NBS1 and MRE11 genes, respectively, cause Nijmegen breakage syndrome (immunodeficiency with microcephaly, chromosomal instability, and a cancer predisposition, but no ataxia114) and AT-like disorder (with neurologic manifestations and radiosensitivity, but no telangiectases).
CLINICAL FINDINGS
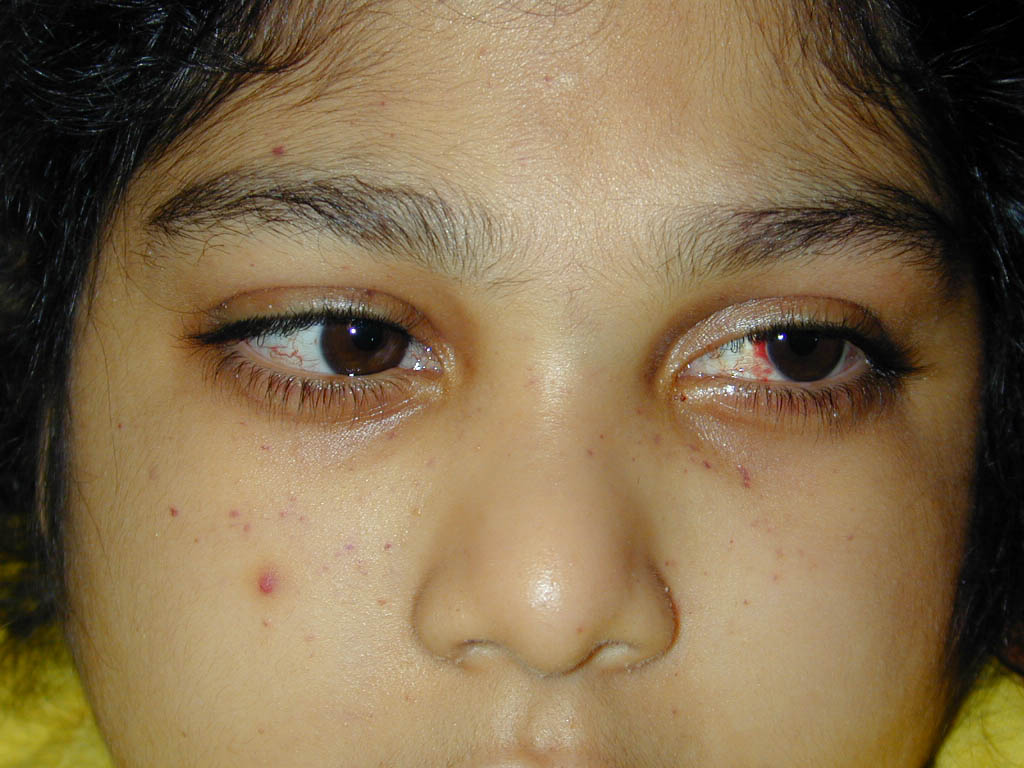
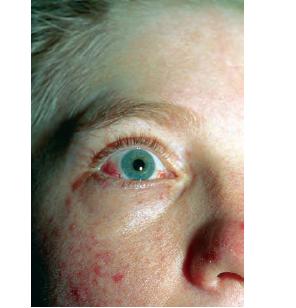
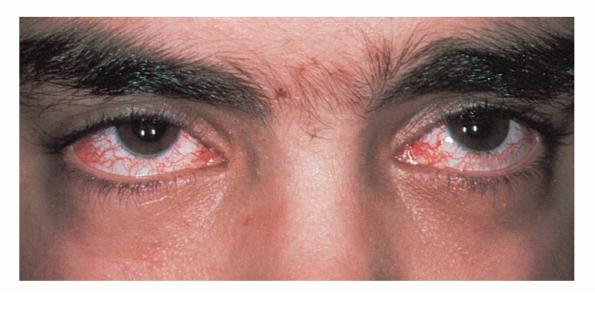
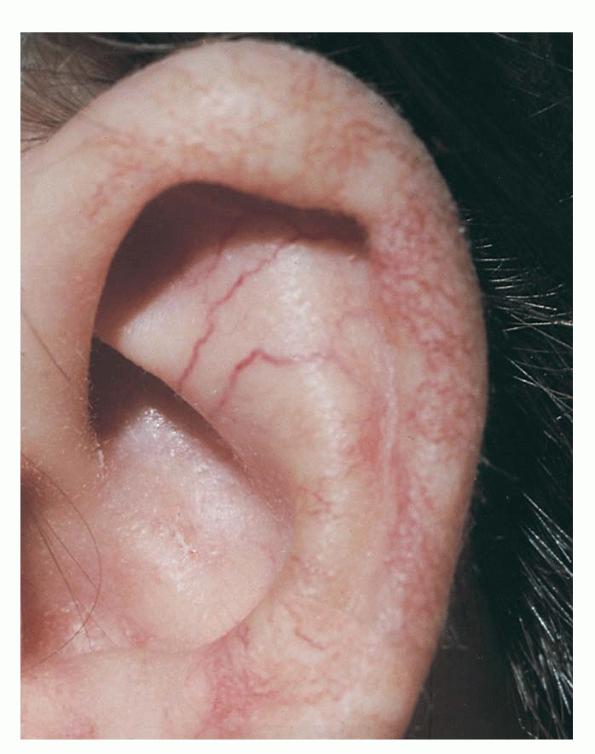
Characteristic oculocutaneous telangiectases begin near the ocular canthi and progress across the bulbar conjunctivae . . These telangiectases
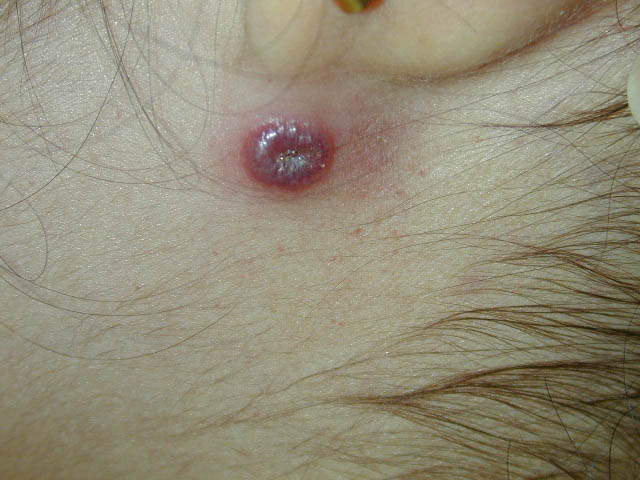
Progeric changes of the skin, including xerosis and gray hair, occur in 90 percent of patients. During adolescence, the facial skin may become progressively more atrophic and sclerotic, causing a masklike appearance. Occasionally, the ears, arms, and hands also become sclerodermatous. The hair may be diffusely gray by adolescence, and subcutaneous fat is generally lost in childhood. Recurrent severe impetigo often develops. Seborrheic dermatitis occurs in many patients, and the associated blepharitis may lead to a diagnosis of blepharoconjunctivitis rather than ocular telangiectasia. Mottled hyperand hypopigmentation commonly occur and, together with the telangiectases and atrophy, can resemble the poikiloderma of radiodermatitis, actinic damage, or scleroderma. Other pigmentary changes include café-au-lait spots that may be found in a dermatomal distribution, multiple ephelides, and vitiligo. Hypertrichosis of the arms and legs, alopecia areata, multiple warts, atopic dermatitis, keratosis pilaris, nummular eczema, and acanthosis nigricans have also been described in association with AT. Among the most common cutaneous manifestations are non-infectious cutaneous granulomas . These persistent, atrophic, and often ulcerative lesions are often mistaken for other granulomatous processes, including sarcoidosis, necrobiosis lipoidica diabeticorum, granuloma annulare, and granulomatous dermatitis. Usually, the progressive cerebellar ataxia first becomes apparent during infancy (median age, 1.2 years old) with swaying of the head and trunk and apraxia of eye movements, often years before skin or conjunctival abnormalities develop. In childhood, dysarthric speech, drooling, choreoathetosis, and myoclonic jerks become prominent. The diagnosis of AT is usually made at a median age of 7 years, after appearance of the mucocutaneous telangiectasia. Patients usually require a wheelchair by their teenage years. Recurrent bacterial and viral sinopulmonary infections occur in up to 80 percent of patients; these are the most common cause of death, which is usually from bronchiectasis and respiratory failure. Approximately 75 percent of patients with AT may have growth retardation and endocrine disorders, especially ovarian agenesis or testicular hypoplasia and insulin-resistant diabetes. Neoplasia occurs in 40 percent of surviving adolescents or young adults, although lymphoid malignancy has been described as the presenting sign during infancy. Most common are lymphomas (especially B cell; 200-fold increased risk) and leukemia (especially T-cell chronic lymphocytic; 70-fold increased risk).118,119 Basal cell carcinomas have been reported in young adults. Among techniques to confirm diagnosis are analysis of radioresistant DNA synthesis (which demonstrates an abnormal S phase checkpoint), radiosensitivity testing with the colony survival assay, immunoblotting for the ATM protein, assessment of ATM kinase activity, and molecular genetic testing.
PROGNOSIS, CLINICAL COURSE
AND TREATMENT
The therapy for AT is supportive and includes administration of antibiotics for infection, physiotherapy for pulmonary bronchiectasis, physical therapy to prevent contractures in patients with neurologic dysfunction, and sunscreens and sun avoidance to diminish actinic-like changes. Patients should be aggressively screened for malignancy, especially after the first decade of life. Intralesional injections of triamcinolone have helped to promote healing of the painful associated ulcerations, although the lesions do not clear completely with treatment. Autopsy findings indicate that approximately 50 percent of the patients die from pulmonary disease, the most common cause of death. Lymphoreticular malignancies, including lymphoid leukemia, are the second most common cause of death, leading to the deaths of 15 percent of patients with AT. The remaining patients tend to die of both pulmonary disease and malignancy. Therapeutic radiation and radiomimetic chemotherapeutic agents, especially bleomycin, may lead to extensive tissue necrosis. The administration of small doses of other chemotherapeutic drugs and low-dose, fractionated radiation is the least harmful means of managing these malignancies. In a small subset of patients with milder AT, treatment with aminoglycosides increased ATM gene function.121 Death usually occurs by late childhood or early adolescence; the oldest surviving patient died at the age of 50 years. Prenatal diagnosis is currently best achieved by DNA analysis.
|